Overview
We are studying the mechanism and regulation of meiotic recombination in mouse. Portions of this work are part of a longstanding collaboration with Maria Jasin’s lab in the Developmental Biology Program at Memorial Sloan Kettering Cancer Center.
Exploring the double-strand break landscape in mouse
DSBs are more likely to occur in some genomic regions than others. Elucidating mechanisms shaping the DSB “landscape” requires identifying the biochemical factors involved, understanding how they influence Spo11, and deconvolving their interactions. We developed a powerful method to address these issues: each Spo11 oligo is a tag recording precisely where a break was made, so deep-sequencing oligos allowed us to quantitatively map DSBs across the yeast genome at nucleotide resolution with high sensitivity. More recently, we extended this methodology to mouse (Lange et al. 2016; Yamada et al. 2017). A major hotspot determinant in most mammals is PRDM9, which sports a histone methyltransferase module and a rapidly evolving Zn-finger DNA binding domain. DNA-binding specificity defines hotspots, but how PRDM9 targets SPO11 activity is unknown. Moreover, although the mammalian DSB landscape is shaped hierarchically over different size scales, as in yeast, studies tend to focus almost exclusively on hotspots so we know little about other features. The much higher resolution and sensitivity of SPO11-oligo mapping compared with prior ssDNA sequencing (SSDS) maps allows us to address many of these challenges in ways not previously impossible.
Role of ATM in feedback control of double-strand break formation
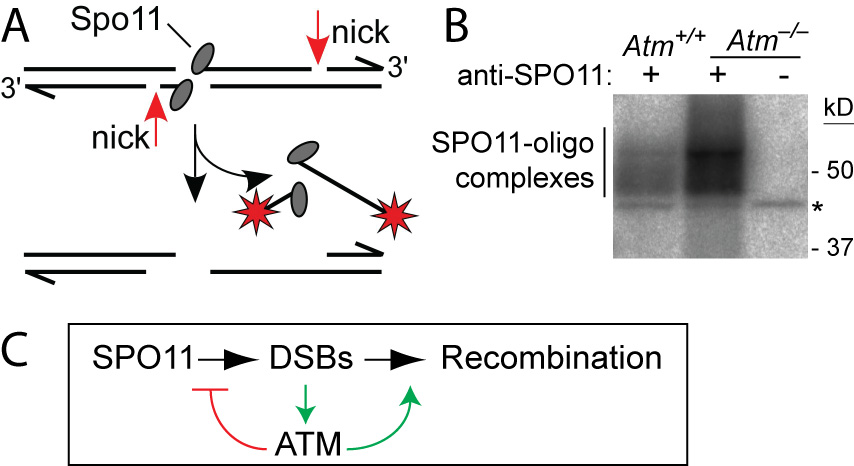
ATM is a Ser/Thr kinase mutated in the cancer-prone disease ataxia telagiectasia (A-T). ATM activated by DSBs triggers cell cycle checkpoints and promotes DNA repair in somatic cells, but ATM is also essential during normal, unperturbed meiosis, for reasons that have been unclear.
A breakthrough came from our discovery that Atm–/– spermatocytes experience greatly elevated numbers of DSBs. Moreover, absence of ATM renders DSB formation extremely sensitive to SPO11 expression level, probably explaining why Spo11 heterozygosity partially rescues Atm deficiency: many defects in Atm-null spermatocytes are likely caused by grossly elevated DSBs, which are lowered by reduced SPO11 expression. We proposed that ATM activation governs a negative feedback loop that restrains SPO11 to limit the number of DSBs (Lange et al., 2011). Independent studies in Drosophila (McKim lab) and yeast (Kleckner, Neale, Cha, and Fung labs) led to a similar conclusion. More recently, we have explored how ATM shapes the DSB landscape in mouse (Lange et al. 2016) and yeast (Mohibullah and Keeney 2017).
X-Y recombination
Sex chromosome segregation poses challenges to the male meiotic cell because the X and Y share only a small region of homology, the pseudoautosomal region (PAR): at least one DSB must form within the PAR; that DSB must locate and engage its homologous partner; and a crossover must form. To understand how cells meet these challenges, we studied the behavior and structure of the PAR in normal meiosis, and explored genetic requirements for ensuring PAR DSB formation (Kauppi et al. 2011). We found that mouse PAR DNA occupies unusually long chromosome axes, potentially as shorter chromatin loops, predicted to promote DSB formation. We also found that most PARs have delayed appearance of the RAD51 foci that mark DSB ends, and we provided evidence that PAR DSBs are genetically distinct from “global” DSBs. These findings uncover specific mechanisms that surmount the unique challenges of X-Y recombination.
Many questions remain, including: What aspects of PAR structure and behavior are intrinsic to the PAR itself? Are pro-DSB factors specifically enriched at the PAR in males? One approach we are using to address these issues is to query by immuno-FISH the higher order structure and protein composition of the PAR in both male and female meiosis. In females, the X chromosomes synapse and recombine along their length like autosomes, and the PAR has a much lower crossover rate than in males. Quantifying PAR RAD51 foci in oocytes will allow us to determine whether the less frequent crossing-over reflects fewer DSBs, a lower likelihood that a DSB becomes a crossover, or both. Measuring axis and loop sizes will test the correlation between chromosome structure and DSB potential. Immuno-FISH will also allow us to test the hy-pothesis that the PAR preferentially accumulates DSB-promoting factors. These and other studies of the PAR compared to autosomes and in spermatocytes compared to oocytes will provide critical information about this important region of the genome and help us understand the mechanisms meiotic cells exploit to promote accurate sex chromosome segregation.
Non-allelic homologous recombination
Recombination usually occurs between allelic sequences on homologs or between sister chromatids, but non-allelic homologous recombination (NAHR) between repetitive sequences can cause chromosome rearrangements. In humans, germ-line NAHR accounts for over 30 types of disease-causing mutation, with phenotypes including mental retardation and male infertility. Little is known about this process: What is the mechanism of meiotic NAHR and how does it differ from allelic recombination? What strategies do cells employ to limit NAHR? From mouse SPO11-oligo maps, we uncovered several repeats containing strong DSB hotspots (Yamada et al. 2017; S. Kim and J. Lange, unpublished data). We are using PCR-based physical assays on sperm DNA to explore the mechanism and regulation of NAHR, comparing allelic and non-allelic recombination frequencies and testing the hypothesis that mismatch repair machinery inhibits NAHR. These studies will provide novel insights into how cells minimize the risk of germ-line genome instability posed by the need to distribute recombination events across a genome that is rife with repetitive elements.
Discovering new meiotic genes in mouse
Aneuploidy (abnormal chromosome numbers) in gametes (sperm or eggs) is the leading genetic cause of miscarriage and birth defects, and is often attributable to chromosome segregation errors during meiosis. Reverse-genetic studies of mouse meiosis have elucidated how meiotic perturbations can cause chromosome segregation errors. However, our molecular understanding of key meiotic processes and their contributions to production of euploid gametes is far from complete. A critical challenge is that we lack knowledge regarding the full complement of mammalian meiotic genes. To address this lack, we are continuing our longstanding effort to identify candidate genes that might be important for meiosis, engineering loss-of-function mutations, and characterizing the reproductive phenotypes of the mutant mice. For example, we defined an unexpected role in spermiogenesis of BAZ1A (a.k.a. ACF1), the defining subunit of the ISWI-family chromatin remodeling complex ACF (Dowdle et al. 2013). Current studies are aimed at understanding ACF function in round spermatids. In current studies we are continuing to identify and mutate additional candidate genes.
Importantly, however, orthology-driven reverse genetics alone will never be capable of cataloging genes that are wholly specific to mammals or genes whose sequences have diverged to the point of obscuring homology with meiotic genes known in model organisms from other taxa. Therefore, we are also employing a phenotype-driven chemical mutagenesis screen in mice to discover novel genes important for germline genome stability. Since the screen involves random point mutagenesis, it also has the potential to generate non-null alleles that reveal unappreciated gene functions. Our screen has picked up genes involved in meiotic recombination, formation of higher order meiotic chromosome structure (e.g., synaptonemal complex (SC)), and/or meiotic sex chromatin inactivation (MSCI). We recently described two novel hits from this screen: Dnmt3c, encoding a previously uncharacterized DNA methyltransferase that is unique to rodents and that controls transposable element expression in the male germline (Jain et al. 2017); and Ythdc2, encoding an RNA helicase that plays a critical role in regulating the switch from mitosis to meiosis in the germline (Jain et al. 2018). In current work we continue to explore the mechanism of action of DNMT3C and YTHDC2, and we are studying additional hits that emerged from the screen.






