High-resolution, genome-wide exploration of the DSB landscape
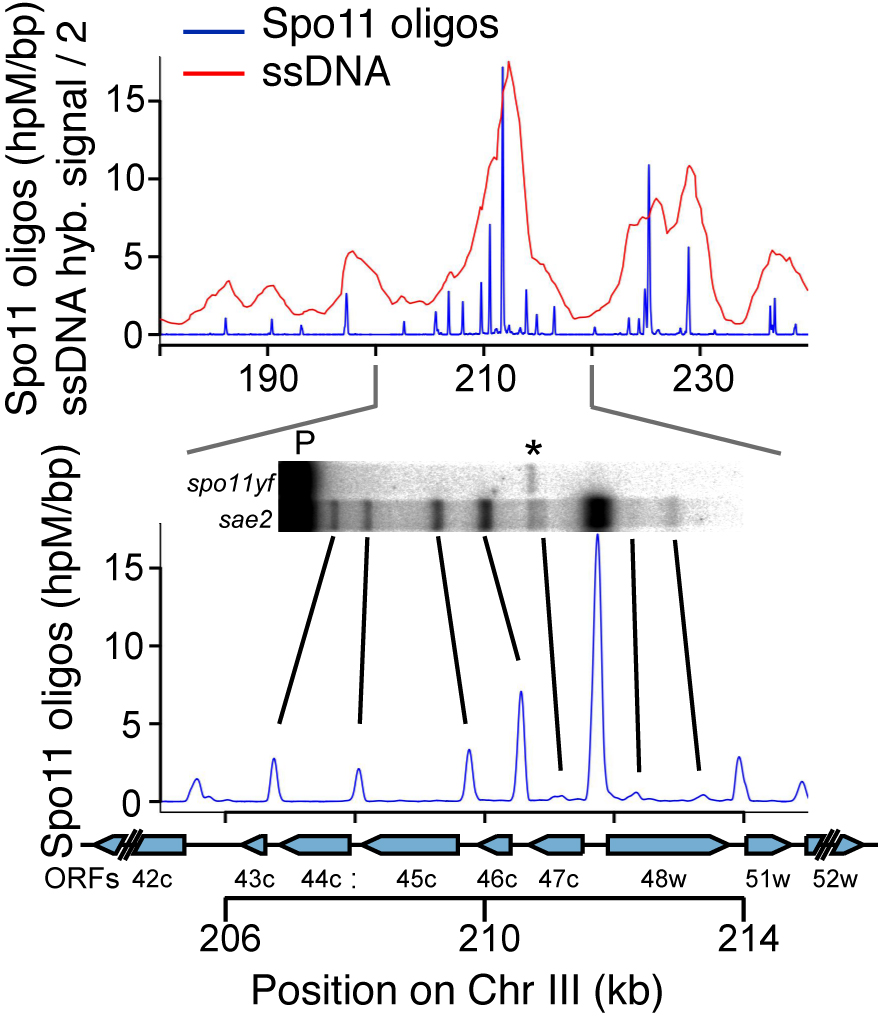
DSBs are more likely to occur in some genomic regions than others. To understand this non-random distribution, we are taking advantage of the Spo11-oligonucleotide complexes formed as a byproduct of DSB formation and processing. Each Spo11 oligo is a tag that records precisely where a break was made. We devised methods to purify, amplify, and deep-sequence the oligos, allowing us to quantitatively map DSBs across the genome at nucleotide resolution with high sensitivity (Pan et al., 2011).
By applying this mapping methodology to wild-type and mutant yeast strains, we are gaining a detailed understanding of the factors that influence DSB distributions (e.g., Zhu et al., 2015; Mohibullah and Keeney 2017). An overarching theme is the view that the DSB landscape is shaped by combinatorial action of many factors. These factors operate hierarchically over many size scales, with the general trend that level in hierarchy is proportional to scale. Thus, for example, two DNA segments that are equally free of nucleosomes may have substantially different DSB probability depending on whether they lie in hotter or colder domains. We have also used Spo11-oligo mapping in various Saccharomyces species to explore the evolutionary dynamics of the DSB landscape (Lam and Keeney 2015), and we have extended Spo11-oligo mapping to S. pombe (Fowler et al. 2014) and mouse.(Lange et al. 2016; Yamada et al. 2017).
Regulating the number, timing, and location of DSBs
DSB formation is a suicide reaction for Spo11 because the protein’s active-site tyrosine remains covalently linked to DNA. In principle, this feature could regulate DSB numbers, but only a small fraction of the protein ever makes a break and Spo11 and its accessory factors remain abundant long after most DSB formation ceases. These features suggest that mechanisms that control DSB numbers work in part by restraining Spo11 activity. Recent work by us and others has uncovered a network of intersecting feedback circuits that control DSB number and distribution. (A) ATM/Tel1. One circuit involves activation of the DSB response kinase ATM (known as Tel1 in yeast). ATM activation triggers a negative feedback loop that inhibits further breakage, presumably via phosphorylation of Spo11-accessory factors (Lange et al. 2011; Mohibullah and Keeney 2017). (B) Homolog engagement. In mice, we found that DSBs continue to accumulate on chromosomes that cannot synapse with a homolog because of pairing defects caused by hypomorphic Spo11 mutation or because no homolog is present (e.g., on the part of X not homologous with Y) (Kauppi et al. 2013). These findings dovetailed with studies of yeast “ZMM” proteins, a suite of factors needed to complete recombination and SC formation. We discovered that ZMM mutants accumulate additional DSBs (Thacker et al. 2014). Putting the mouse and yeast results together, we proposed that homologous chromosomes that successfully engage one another stop making DSBs. A plausible mechanism is that SC formation leads to structural changes that render chromosomes unfit substrates for Spo11. (C) Temporospatial coordination of DSB formation with prior DNA replication. It has been long appreciated that during meiosis DNA replication is coordinated with the subsequent formation of DSBs. We found that in yeast the replisome-associated components Tof1 and Csm3 physically associate with the Dbf4-dependent Cdc7 kinase (DDK) and recruit it to the replisome, where it phosphorylates the DSB-promoting factor Mer2 in the wake of the replication fork, synchronizing replication with an early prerequisite for DSB formation (Murakami et al. 2014). We speculate that recruiting regulatory kinases to replisomes may be a general mechanism to ensure spatial and temporal coordination of replication with other chromosomal processes. (D) Coordination with meiotic progression. Pachytene exit in yeast is controlled by the Ndt80 transcription factor. Recombination products and DSBs accumulate in ndt80 mutants, leading to the proposal that pachytene exit ends a period permissive for DSB formation, i.e., that Ndt80 is an indirect negative regulator of Spo11. Confirming this hypothesis, multiple lines of evidence now show that ndt80 mutants make more DSBs. Interestingly, recombination defects inhibit Ndt80 activation via checkpoint responses mediated by Mec1 kinase (yeast ortholog of mammalian ATR), thus establishing complex regulatory circuits connecting DSB repair to the potential to make additional DSBs. By epistasis analysis, this Ndt80 circuit is genetically distinct from feedback through homolog engagement.
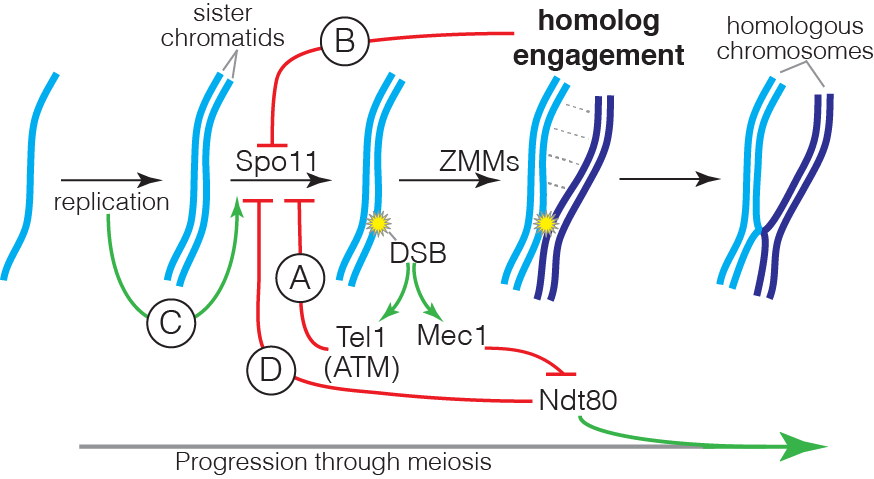
Intersection of the DSB control circuits creates potential for complex compensatory interactions. For example, ZMM mutations likely impinge on multiple circuits simultaneously, removing restraints on Spo11 activity by disrupting homolog engagement and inhibiting Ndt80 activation, but also hyperactivating negative feedback via Tel1. The detailed mechanisms of DSB regulation by ATM/Tel1 and by homolog engagement are currently unknown, and are a key target of present and planned investigation. Deriving a nuanced understanding of the control of Spo11 requires us to account for the biological complexities within the DSB control network; achieving this goal is a critical element of our experimental design.
Mechanism of DSB resection
Meiotic DSBs are processed by 5’-to-3’ exonucleolytic resection to generated single-stranded DNA tails for recombination. Resection occurs through a two-step process in which endonucleolytic cleavage by the Mre11-Rad50-Xrs2 complex plus the Sae2 protein generates nicks on Spo11-bound strands that serve as entry points for modest 3’-to-5’ exonuclease activity of Mre11 chewing back toward Spo11, plus robust 5’-to-3’ exonuclease activity of Exo1 chewing away. This resection is a conserved feature of recombination but remains poorly understood. We developed a next-generation-sequencing method to map resection endpoints genome wide. This method, S1-seq, involves removal of ssDNA with S1 nuclease and ligation of sequencing adaptors to the blunted DNA ends. Using this method, we explored the patterns and mechanisms of DSB resection in yeast (Mimitou et al. 2017). This work uncovered roles of Tel1 kinase in regulating resection, and revealed intriguing interactions between the resection machinery and the chromatin structure of the broken chromosomes. Current work aims to understand the role of chromatin in shaping meiotic resection. We are also extending these studies by adapting S1-seq to use in mouse meiosis.
Spore-autonomous fluorescent protein reporters

Studies of meiotic recombination often rely on microdissecting the four products of a single meiosis and analyzing the configuration of genetic markers in viable spore clones. This type of analysis is powerful but laborious, and reliance on viable spores may introduce selection bias. To overcome these limitations, we developed new markers whose segregation can be scored without ascus dissection, even in inviable spores. Using previously described promoters that can drive fluorescent protein expression specifically in only those spores that inherit the reporter, we developed a versatile, portable set of “spore-autonomous” reporters that can be integrated essentially anywhere in the genome. Proof-of-principle experiments showed that different configurations can be used to quantify crossover frequency, crossover interference, gene conversion, crossover/noncrossover ratios, and chromosome missegregation.





