Despite the importance of DNA replication, numerous aspects of this process are still poorly understood. One fundamental question is: how do replication forks efficiently progress through chromatin? Understanding this question is complicated by the diversity of chromatin structure and the heterogeneity of replication reactions.
The analysis of nucleic acids by high-throughput sequencing has dominated biological research for more than a decade. Seminal studies that precisely map the production of nascent RNA (Gro-seq) at elongating RNA polymerase II (Net-seq); or RNA in the process of translation (Ribosome-profiling) have revolutionized our ability to interrogate RNA metabolism with high resolution. However, the study of DNA replication at nucleotide resolution with genomics has significantly lagged the transcription/RNA fields. This is principally because nascent DNA molecules are limited in copy number, differ in length by several orders of magnitude and are difficult to unambiguously separate from their template. As a result, assays analogous to those which are commonly used to map nascent RNA cannot be performed for DNA. Such limitations have significantly hampered our ability to understand how the replication machinery progresses through chromatin and how replication forks are processed when they are stalled by impediments such as DNA damage.

Most genomics methods that study DNA replication rely on population-based assays; while these are powerful, they miss infrequent or stochastic events that occur within the mixture of cells. Such rare events, such as fork stall or collapse, are highly biologically significant, but we understand little of where and when they may occur across a genome. To overcome these limitations, we have recently developed a new genome-wide approach to capture DNA replication with single event readouts.
Our new method – Replicon-seq – extracts and sequences DNA that has been synthesized by two replisomes that originated from the same origin of replication (replicon). We reasoned that creation of DNA strand breaks at functional replisomes would allow the position of any replisome to be identified by mapping the ends of nascent DNA by sequencing. Moreover, the relative positions of sister replisomes can be identified if nascent DNA chains could be sequenced in their entirety.
We chose to fuse Micrococcal Nuclease (MNase) to the replicative helicase, CMG, which is likely the most stable component of the replisome. We performed experiments in which S. cerevisiae are arrested in G1 with alpha-factor and the thymidine analogue BrdU is added to allow labeling of nascent DNA upon release into S-phase. Cells were harvested in early S-phase, permeabilized and MNase activated with the addition of calcium for limited time. The DNA is directly sequenced using nanopore technology with the output expected to be composed of nascent – BrdU containing molecules – as well as parental template and un-replicated DNA. BrdU containing reads which represent nascent molecules are identified by informatically due to current shifts during nanopore sequencing.
Nanopore sequencing of replicons proceeds from the 5’ end of the lagging strand through to the 3’ end of the leading strand of the sister replisome. Thus, the ends of replicons provide a high-resolution, single-molecule readout of the relative position of sister replisomes along the genome. Plotting individual BrdU-containing reads according to genomic location and length reveals highly symmetric patterns which emanate from known replication origins.
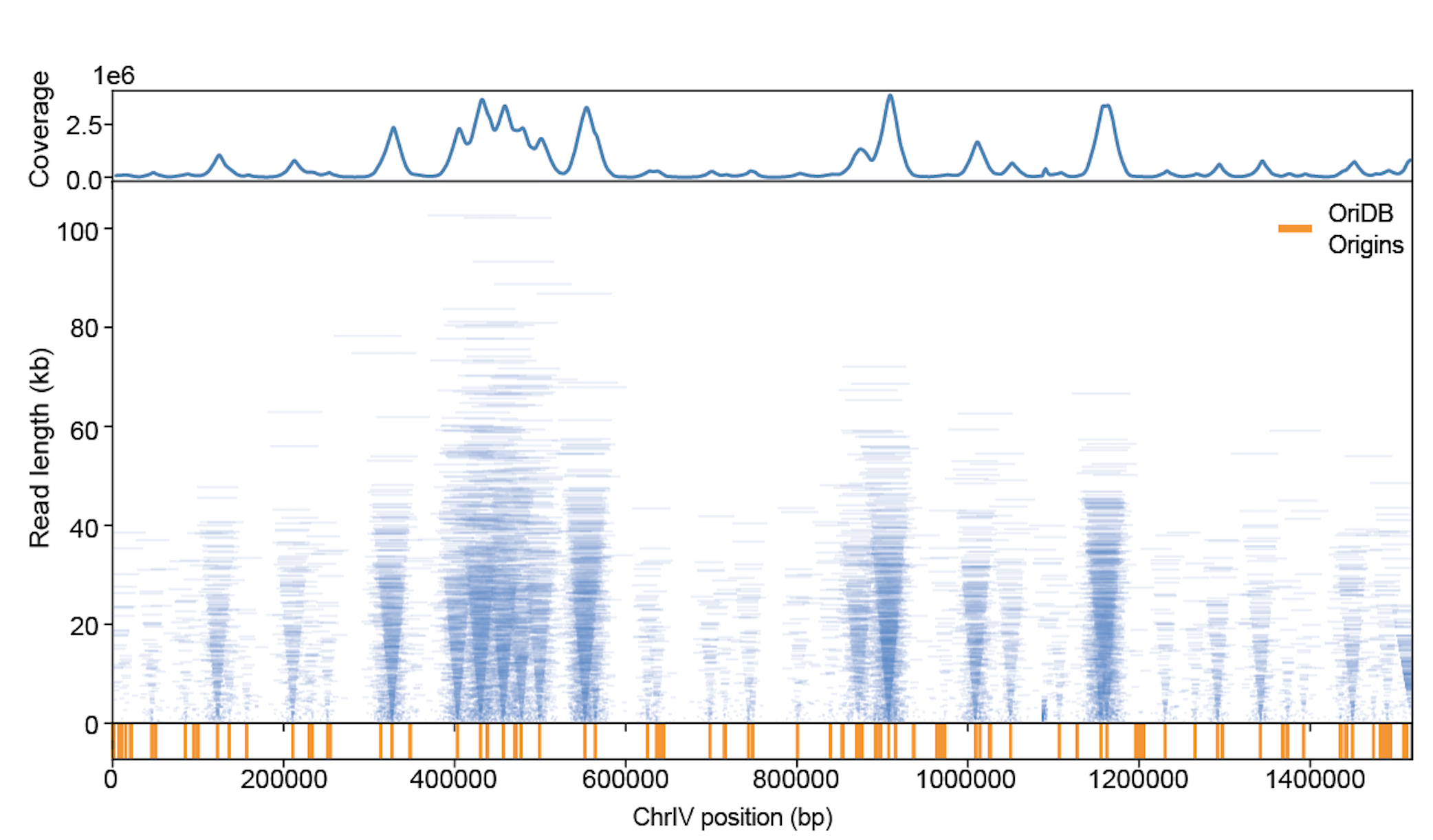
We are currently utilizing replicon-seq to study several important aspects of DNA replication, such as: how the replication machinery deals with impediments such as gene transcription, how replication termination is achieved and how nucleosomes impede replication fork progression.