Alveolar rhabdomyosarcoma is a cancer of the muscle lineage. Cancer Cachexia is a muscle and fat wasting syndrome associated with cancers of the pancreas, lung, colon. Nemaline myopathy is associated with defects in sarcomere structure and function. We are investigating disease progression and performing screens to identify novel therapeutics for these diseases.
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is a rare tumor, yet it is the most common pediatric soft tissue sarcoma, representing 3 to 5 percent of all childhood cancers. Patients with alveolar rhabdomyosarcoma (ARMS), an aggressive form of RMS, often have poor prognosis, particularly with metastatic disease. There are no targeted treatments for this cancer.
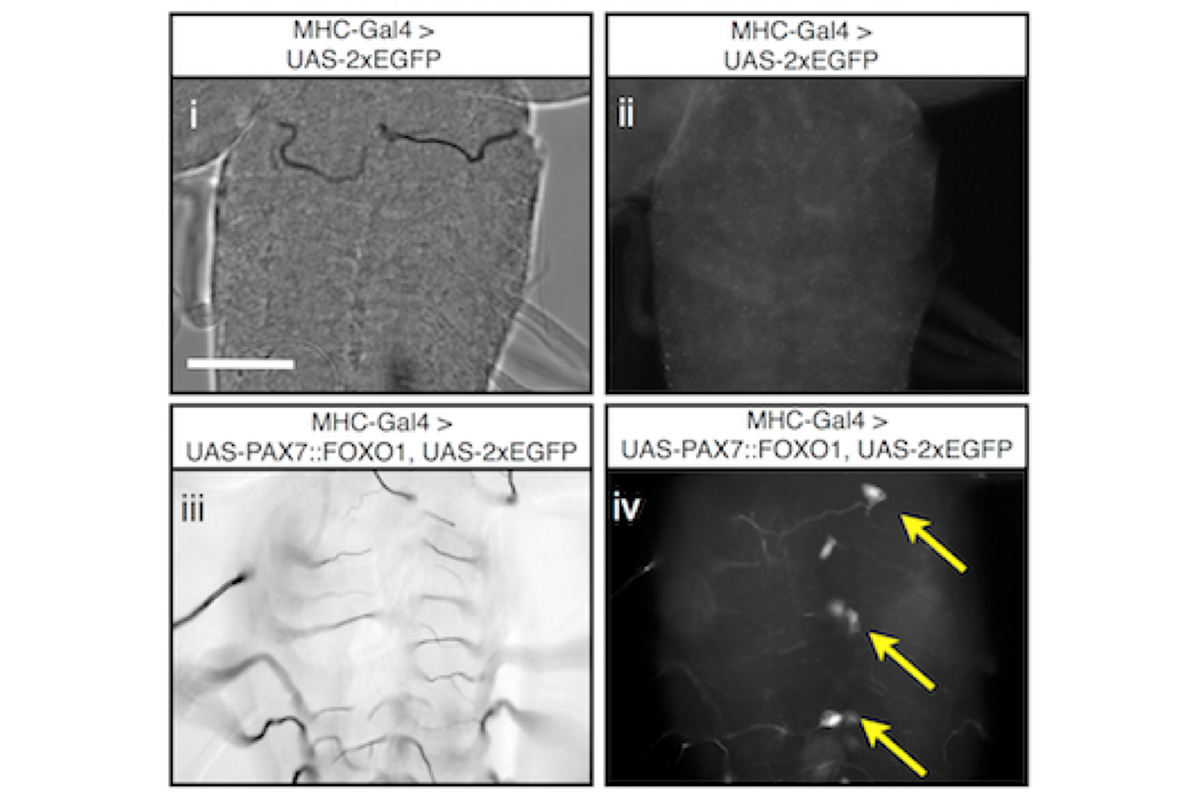
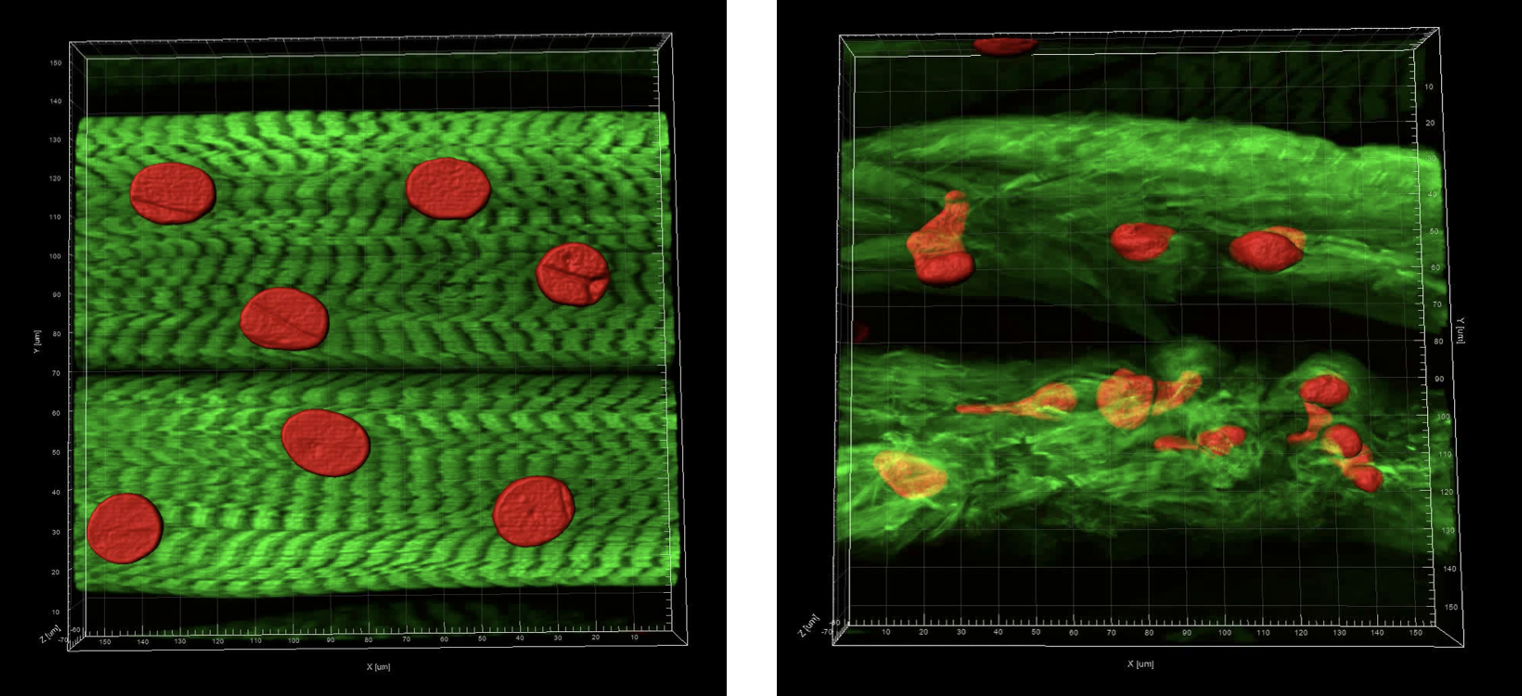
ARMS uniquely associates with chromosomal translocations t(2;13)(q35;q14) and t(1;13)(p36;q14), leading to the production of the fusion oncoproteins PAX3::FOXO1A or PAX7::FOXO1A. Expression of a human PAX7::FOXO1A fusion protein alone in Drosophila mesoderm leads to a population of free larval myoblasts that transform into aggressive cells that leave the muscle compartment, migrate, and invade nonadjacent tissues, including the central nervous system (CNS).
This cellular behavior is reminiscent of metastatic disease in which transformed cells invade and integrate into adjacent tissues. We are studying this Drosophila ARMS model to better understand metastatic behaviors. A screen for new therapeutics to treat this disease is underway. We are currently translating our screen positives to human cancer cells and mouse xenograft studies.
Cancer Cachexia
The cancer-associated cachexia syndrome (CACS) is a systemic metabolic disorder that is characterized by catabolism of stored nutrients in skeletal muscle and adipose tissue. This syndrome is particularly prevalent in patients with pancreatic, lung, and colon cancers and contributes to poor quality of life, treatment toxicity, and increased mortality. While several systemic factors have been implicated in CACS, there continues to be no definitive mechanism of action and no FDA-approved therapy.
A glaring omission from the existing literature base is a description of the early and progressive changes that occur in skeletal muscle prior to the onset of severe wasting. Therefore, current projects are aimed at addressing this critical gap in knowledge by delineating the fundamental mechanisms driving atrophy in human muscle. To this end, we are using both Drosophila and human muscle organoids as model systems. Our goal is to understand and then design therapeutics to prevent such wasting.
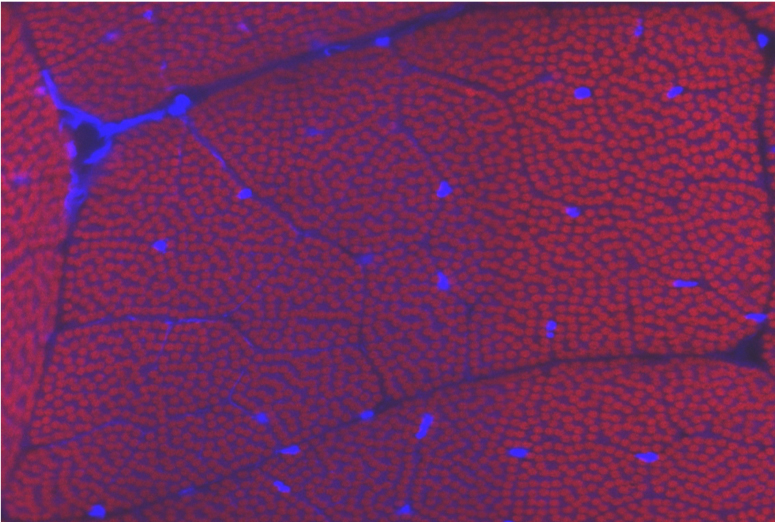
Nemaline myopathy
Nemaline myopathy (NM) is one of the most common form of non-dystrophic congenital myopathy, with an incidence of 1/20,000 in some populations. It is a slow or non-progressive disease, characterized by the presence of nemaline bodies in the affected muscle tissue, muscle weakness, and the absence of muscle regeneration. There is no effective treatment for this disease.
Mutations in over nine different genes have been linked to NM, six of which encode skeletal muscle sarcomere thin filament-associated proteins. Using the human mutations as a guide, we have developed Drosophila models for the disease. We are currently investigating how NM develops and are identifying novel therapeutics to treat it. We are translating these studies to our human muscle models.