Our laboratory is interested in how human cells accurately duplicate and transmit their chromosomes during each round of division.
Accurate chromosome transmission is vital for proper embryogenesis and renewal of healthy tissues in the adult. Conversely, defects in duplicating and segregating chromosomes are associated with infertility, birth defects, and cancer. Chromosome transmission errors may also sensitize tumor cells to specific kinds of therapies that attack this trait as an Achilles’ heel. To illuminate these issues, our lab has developed and refined methods for precisely mutating genes in human somatic cells via homologous recombination. Using these genetic model systems, we have discovered a number of novel regulatory mechanisms that couple chromosome and cytoskeletal dynamics with cell cycle progression, thus ensuring genome stability.
Genome Engineering and Chemical Genetics in Human Cells
Over the past decade, we have established a robust technology platform for introducing precise genetic alterations (i.e., conditional or constitutive “knockout” or “knockin” mutations) into human somatic cells. Our published protocols take advantage of adeno-associated virus (AAV) vectors, which are delivered to the nucleus as single-stranded DNA and thus require no additional processing to engage the homologous recombination machinery (Berdougo et al., 2009). As such, AAV-mediated gene editing is highly efficient (up to 20 percent targeting efficiency, or about a hundred-fold better than linearized double-stranded DNA) and has been especially fruitful in combination with kinase-engineering strategies developed by Kevan Shokat’s group, whereby a given kinase’s catalytic pocket is enlarged to accept bulky ATP analogs as allele-specific inhibitors.
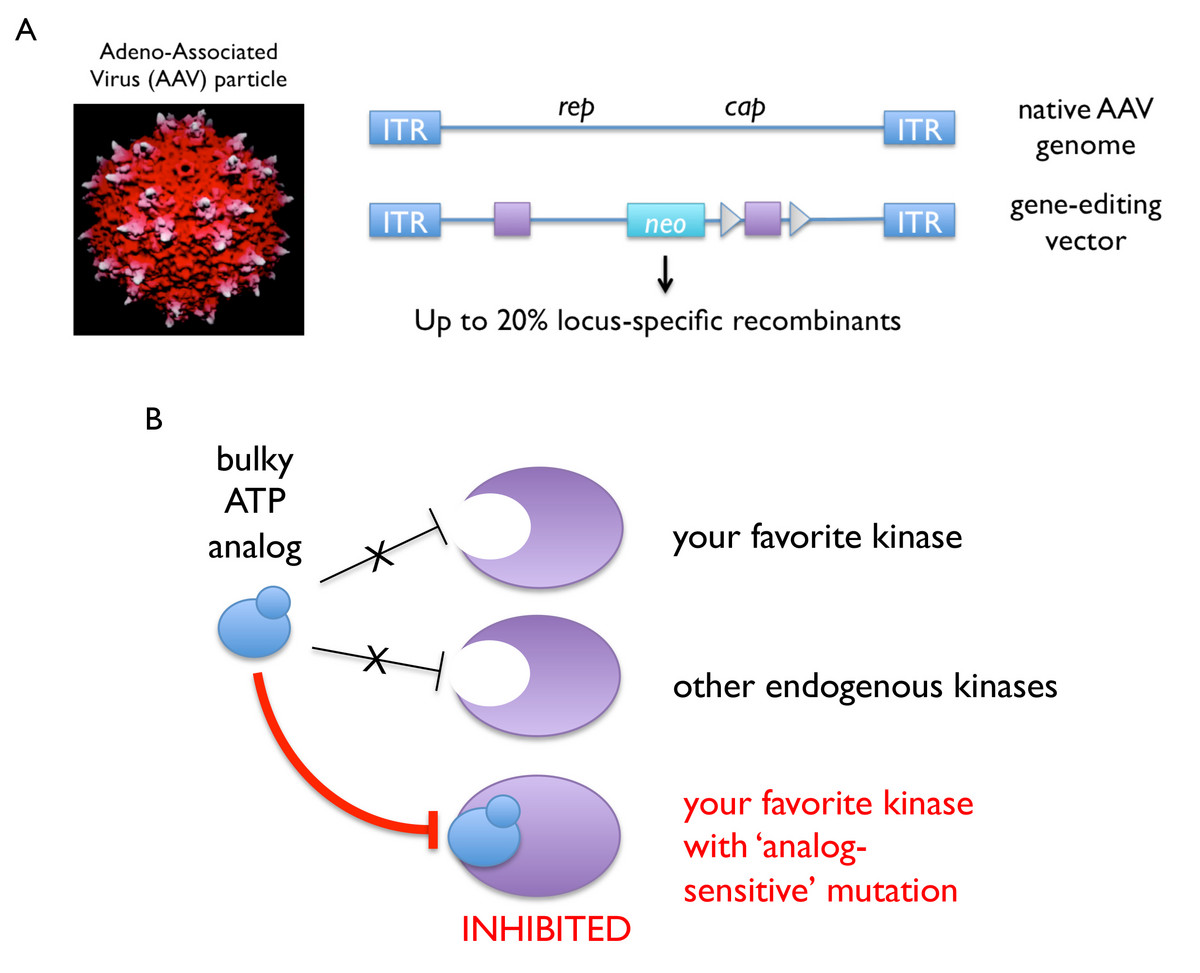
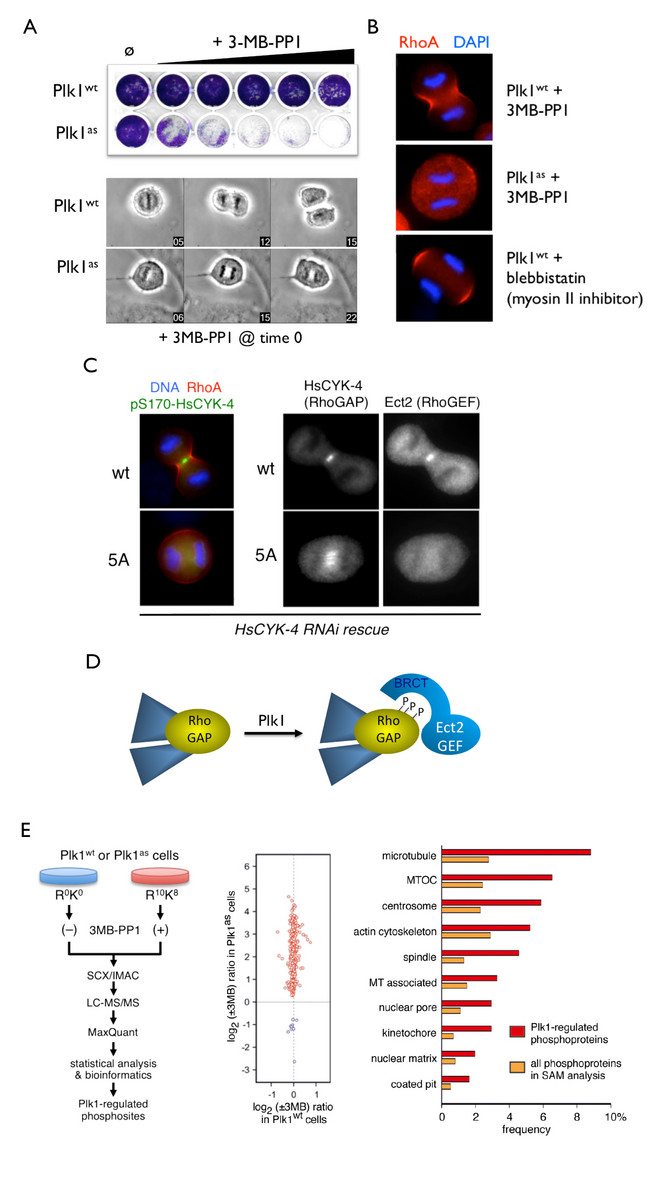
Using this approach, we created human cells in which the endogenous Polo (Plk1) and Mps1 kinases were deleted from the genome and replaced with analog-sensitive variants. The high selectivity and temporal control of these chemical-genetic systems were crucial in revealing new roles of each kinase throughout the cell cycle. For instance, acute inhibition of Plk1 at anaphase onset revealed its essential requirement for targeting the RhoGEF Ect2 to the central spindle, activating the RhoA GTPase at the cell equator, and triggering the onset of cytokinesis (Burkard et al, 2007; Burkard et al., 2009). In collaboration with Bryan Tsou’s group in the Cell Biology Program at the Sloan Kettering Institute, we used similar approaches to show that Plk1 and the cohesin protease separase act sequentially to disengage centrioles at mitotic exit, thereby synchronizing the centrosome and chromosome cycles (Tsou et al., 2009).
Likewise, our chemical-genetic studies on Mps1 revealed that this kinase transduces a “wait anaphase” signal throughout the cell cycle, rather than in strict coordination with outer kinetochore assembly (Maciejowski et al., 2010). Furthermore, our analog-sensitive cell lines have proven to be exquisitely specific and powerful tools for substrate discovery (Oppermann et al., 2012). We are now extending this approach to numerous other kinases, using both AAV vector technology and CRISPR/Cas9 RNA-guided nucleases.
Establishing Sister Chromatid Cohesion during DNA Replication
Another area of emphasis is the problem of how sister chromatids are paired during their synthesis at the replication fork. This process, called sister chromatid cohesion, is required not only for accurate chromosome segregation but also for error-free repair of double strand DNA breaks in post-replicative cells. We discovered that the machinery responsible for cohesion establishment also affects how sister DNAs themselves are synthesized, as deficiencies in the former (including two key acetyltransferases that modify cohesin’s Smc3 subunit, as well as the cohesion-specific replication factor C (RFC)-Ctf18 complex) adversely affect the speed and processivity of DNA replication forks, as shown through DNA combing studies (Terret et al., 2009).
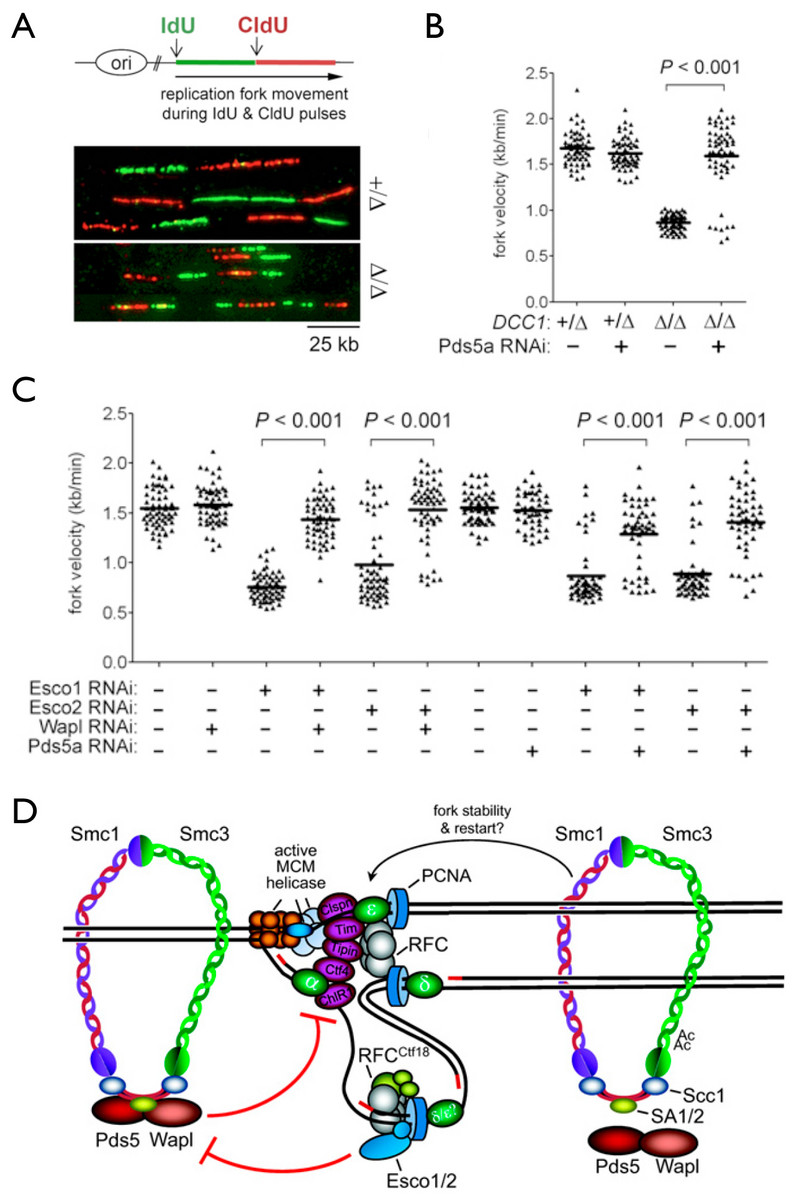
We are presently using genetic, biochemical, and cell biological approaches to determine how this coordination is implemented both spatially and temporally. Of note, a number of inborn developmental disorders (termed “cohesinopathies”) are associated with defects in cohesin itself, the factors involved in cohesion establishment, or proteins that load cohesin onto chromatin (Sherwood et al., 2010). In addition, mutations in cohesin are increasingly seen in cancer genome sequencing. These and other data point to an important role of cohesin in regulating overall chromatin organization and gene expression. Thus, another area of active inquiry in the lab centers on how cohesion regulators affect chromosome architecture, as well as the structure and function of replication and transcription “factories” within the interphase nucleus.
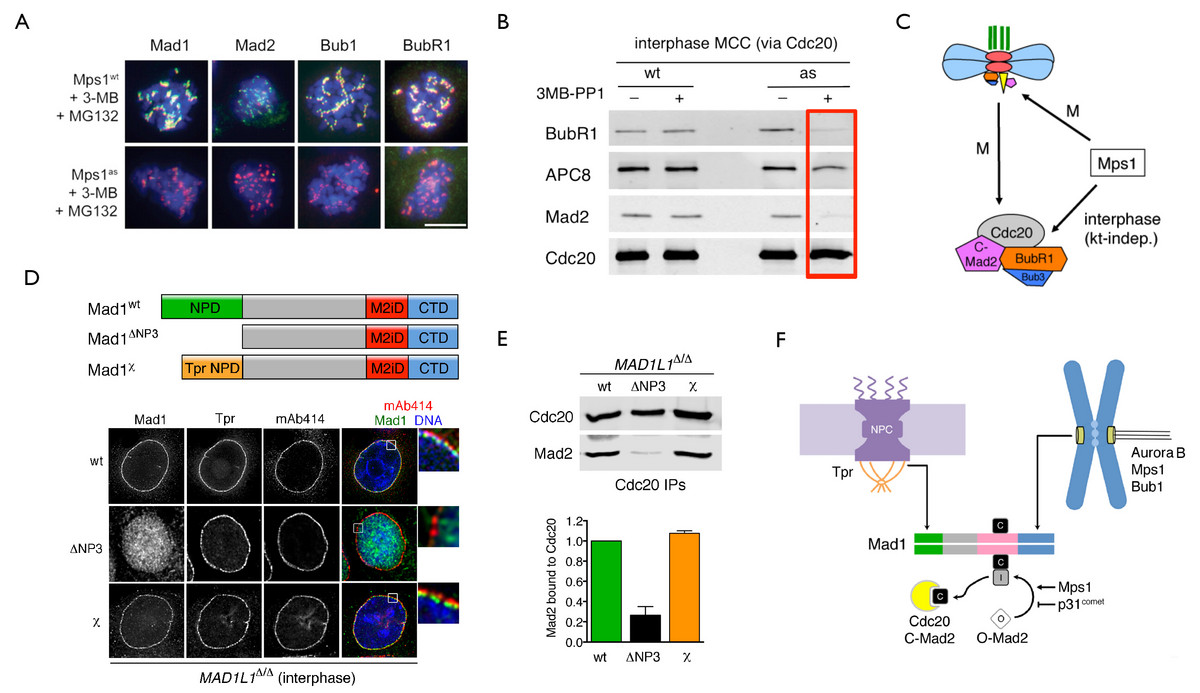
A Novel Role of Interphase Nuclear Pores in the Mitotic Checkpoint
In addition to genome engineering and chemical genetics, we make extensive use of quantitative high-resolution microscopy on both fixed and live cells, as well as mathematical modeling in collaboration with computational biologists. This effort led to the discovery of a novel mode of signal transduction in interphase cells that dictates the speed and fidelity of the subsequent mitosis. This regulation requires both the Mps1 kinase (Maciejowski et al., 2010) and Mad1-Mad2 heterodimers bound to nuclear pore complexes (NPCs; Rodriguez-Bravo et al., 2014). Together these factors trigger the formation of a potent anaphase inhibitor known as the mitotic checkpoint complex (MCC) ahead of M phase entry. This premade MCC pool not only sets the basal timing of an unperturbed mitosis but also increases the sensitivity of the canonical spindle assembly checkpoint (SAC) or mitotic checkpoint response transduced by unattached kinetochores, and also aids in the correction of mitotic errors (specifically merotelic attachments) that are not under SAC surveillance. Insights into this problem had been elusive due to the limited penetrance and specificity of earlier methods, underscoring how newer and more precise technologies can drive major paradigm shifts. We anticipate that continued application of these approaches will illuminate the circuitry that allows our cells to divide accurately, and also inform us of novel strategies for eliminating those cells that fail to do so.