The Lai lab uses an integrated approach to study selected topics in mRNA processing and modification, including alternative polyadenylation, circularization and RNA methylation.
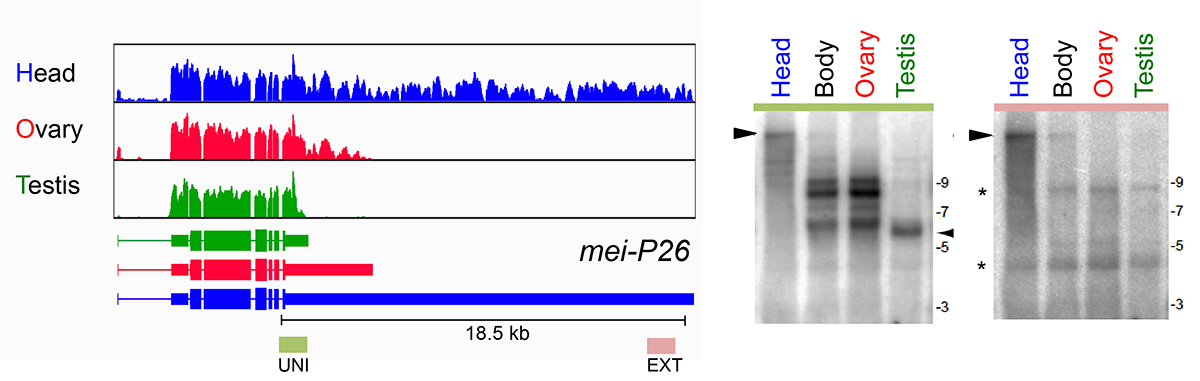
Tissue specific alternative polyadenylation (APA)
As a core member of the modENCODE project, we were deeply involved in reannotating the transcriptome of Drosophila melanogaster, including its short RNAs and conventional transcriptome. One of the surprises of the latter effort was the realization that hundreds of genes utilized unannotated 3’ UTR extensions in the nervous system, and/or shorter 3’ UTRs in the testis. These tissue-specific alternative polyadenylation (APA) programs radically change how we view the post-transcriptional landscape of different settings in the animal. In recent efforts, we have extended this perspective by generating dozens of 3’-seq libraries across development, tissues, and cell lines, and across multiple Drosophila species. These data provide an unprecedented view into
What mechanisms drive tissue-specific APA programs, in cis and in trans? We are analyzing this using bioinformatic queries of our extensive datasets, coupled with molecular genetic screening using the Drosophila model. We are also quite interested to understand the biological imperatives for tissue specific APA. Presumably this reflects needs to alter post-transcriptional regulatory networks. We are examining whether this has impact on specific miRNAs that we have genetic knowledge of. Perhaps more interesting is the genetic perturbation of 3’ UTRs in vivo using CRISPR engineering, to reveal potential phenotypes upon their deletion.
RNA circularization
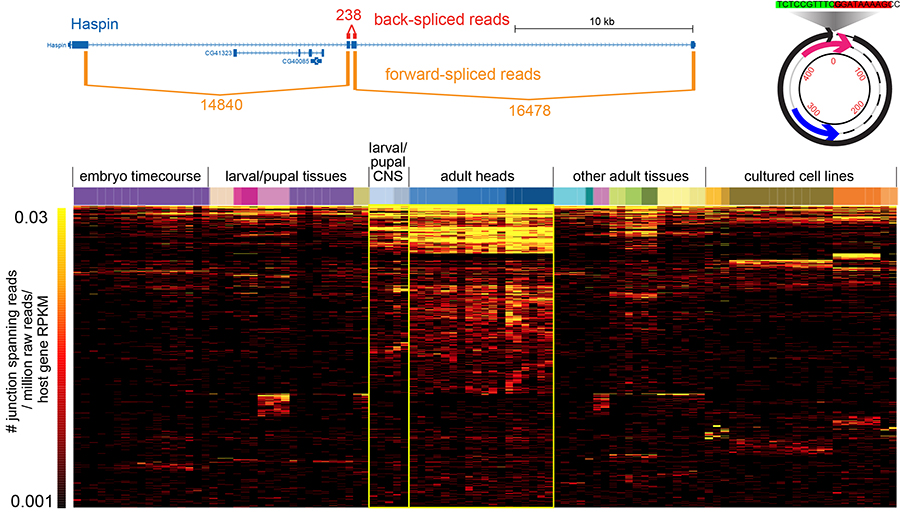
Circularization was recently recognized to broadly expand transcriptome complexity. We exploited massive Drosophila total RNA-sequencing data (>5 billion paired-end reads from >100 libraries covering diverse developmental stages, tissues and cultured cells) to rigorously annotate >2500 fruitfly circular RNAs (Figure 2). These mostly derive from back-splicing of protein-coding genes and lack poly(A) tails, and circularization of hundreds of genes is conserved across multiple Drosophila species. We elucidate structural and sequence properties of Drosophila circular RNAs, which preferentially derive from neural genes and exhibit enhanced accumulation in neural tissues. Interestingly, circular isoforms increase dramatically relative to linear isoforms during CNS aging, and constitute a novel aging biomarker. We are interested to analyze more about the mechanisms of circularization and potential biological activities.
RNA methylation
Of the ~100 post-transcriptional chemical modifications to RNA known, N6-methyladenosine (m6A) is the most prevalent modification in mRNA. Although described over 40 years ago, understanding the biological roles of m6A during most of this time was limited by lack of knowledge of molecular mechanisms that generate and interpret this modification, uncertainty on identities of methylated transcripts, and paucity of genetic mutants in specific m6A pathway components that could reveal in vivo requirements of this nucleoside modification. However, recent years have witnessed remarkable progress on all of these fronts, and the realization that m6A is a major epitranscriptomic modification.
Since most studies of this pathway have been conducted in cultured cells, we established the Drosophila as a powerful system that can combine genetics, biochemistry, and genomics to elucidate in vivo biology of RNA methylation. We used miCLIP to map m6A across embryogenesis, characterize its m6A “writer” complex and YTH readers, and used CRISPR/Cas9 to mutate all the m6A factors. While m6A factors with additional roles in splicing are lethal, m6A-specific mutants are viable but present certain developmental and behavioral defects. Notably, we find that m6A working through the nuclear reader YT521-B facilitates sex-specific splicing of the master female determinant Sxl (Figure 3). In the future, we are interested to use these molecular and genetic toolkit to elucidate other aspects of in vivo m6A biology.

Lee S, Chen YC, FCA Consortium, Gillen A, Taliaferro JM, Deplancke B, Li H and Lai EC (2022). Diverse cell-specific patterns of alternative polyadenylation in Drosophila. Nature Communications.
Joseph B, Scala C, Kondo S and Lai EC (2022). Molecular and genetic dissection of recursive splicing. Life Science Alliance 5: e202101063.
Kan L, Ott S, Joseph B, Park ES, Dai W, Kleiner RE, Claridge-Chang A and Lai EC (2021). A neural m(6)A/Ythdf pathway is required for learning and memory in Drosophila. Nature Communications 12, 1458.
Lee S, Wei L, Zhang B, Goering R, Majumdar S, Wen J, Taliaferro JM and Lai EC (2021). ELAV/Hu RNA binding proteins determine multiple programs of neural alternative splicing. PLoS Genetics 17, e1009439.
Joseph B and Lai EC (2021). The Exon Junction Complex and intron removal prevents re-splicing of mRNA. PLoS Genetics 17(5):e1009563.
Wei L, Lee S, Majumdar S, Zhang B, Sanfilippo P, Joseph B, Miura P, Soller M and Lai EC (2020). Overlapping Activities of ELAV/Hu Family RNA Binding Proteins Specify the Extended Neuronal 3’ UTR Landscape in Drosophila.Molecular Cell 80, 140-155.
Garaulet DL, Zhang B, Wei L, Li E and Lai EC (2020). miRNAs and Neural Alternative Polyadenylation Specify the Virgin Behavioral State. Developmental Cell 54, 410-423.
Joseph B, Kondo S and Lai EC (2018). Short cryptic exons mediate recursive splicing in Drosophila. Nature Structural and Molecular Biology, 25: 365-371.
Westholm, JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR and Lai EC (2014). Genomewide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Reports 9: 1966-1980.