The Overholtzer lab studies mechanisms of nutrient sensing and cellular responses to nutrient starvation, including the nutrient recycling pathway autophagy, scavenging pathways such as macropinocytosis, and mechanisms of cell death. We investigate cell death mechanisms that have unique effects on cell populations, including entosis that supports the survival of starved cells and promotes cell competition, and ferroptosis that eliminates starved cell populations by propagating from cell to cell. The lab also studies mechanisms that regulate autophagy protein and lysosome function in endocytic trafficking and nutrient homeostasis.
Cell Death Mechanisms Induced by Nutrient Starvation
Cells experiencing deprivation of key nutrients such as glucose, amino acids, and oxygen face challenges to support metabolism and to survive. Cancer cells in particular are often nutrient-deprived due to insufficient vasculature in the tumor microenvironment. The uncontrolled proliferation of cancer cells also places a heavy demand on metabolic pathways, leading to exhaustion of available nutrients. We study how starvation affects cell survival, how cell death pathways contribute to cancer progression or therapy, and how cancer cells adapt to starvation by scavenging alternative nutrient sources.
Entosis: Non-apoptotic Cell Death that Promotes Cell Competition

Although it was once thought that most programmed cell deaths occurred by the caspase-dependent program apoptosis, now numerous non-apoptotic programs have been identified in a rapidly expanding alternative cell death field. We have contributed to this field by discovering an alternative mechanism called entosis (Fig. 1) and uncovering its regulation and effects on cell populations. By entosis, cells utilize the machinery of cell adhesion, autophagy, and lysosomes to engulf and kill their live neighbors, in a caspase-independent manner. Entotic cell killing involves autophagy proteins that facilitate lysosome fusion to the endosomes surrounding engulfed cells, an activity that is part of an emerging field of non-canonical autophagy called LC3-Associated-Phagocytosis (LAP).
We have shown that entosis occurs in cancers, and that this process inhibits transformed growth, suggesting that entosis can act as a mechanism of tumor suppression. Conversely, entosis also promotes the development of aneuploidy and allows engulfing cells to scavenge nutrients. These activities may promote tumor progression in the long term. This dual nature of enosis reads out as a form of cell competition, where the engulfment and killing of “loser” cells by neighboring “winners” in a population promotes selection for cells with aggressive characteristics, including increased rates of aneuploidy, altered mechanical properties, and metabolic advantages (Fig. 1).
In recent studies we have found that entosis is induced in cancer cells as a long-term response to starvation. Long-term glucose starvation induces high levels of entosis and selects for winner cells that have ingested, in some cases, many neighboring losers (Fig. 2). Winner cells utilize the nutrients scavenged from losers to grow and proliferate, leading to the outgrowth of cells that are selected for the ability to ingest their neighbors at high rates.
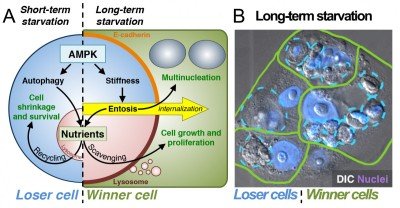
Figure 2. Entosis is induced by long-term starvation. (A) Short-term starvation for glucose induces AMPK-dependent autophagy and nutrient recycling (left). Long-term glucose starvation induces AMPK-dependent entosis (right), which feeds neighboring winner cells that scavenge nutrients. (B) After 72 hours of glucose starvation, winner cells (green) contain one or many ingested losers (blue); nuclei are shown in purple.
Starvation-induced entosis results from increased activity of the energy-sensing kinase AMP-activated protein kinase (AMPK) in loser cells. AMPK signaling promotes an increase in cell stiffness that drives entotic ingestion into winners (Fig. 2). AMPK signaling is also well known to activate the nutrient recycling mechanism autophagy. We have therefore found that starvation signaling can induce two distinct cellular responses, to promote the survival of individual cells in the short-term through autophagy or to promote the outgrowth of selected cells in a population in the long term through entosis. In developing tissues, cell competition is known to induce the outgrowth of winner cells at the expense of “less fit” losers to promote overall tissue fitness. Our data have uncovered a long-term starvation response that may act similarly in cancers. By mobilizing nutrients to winner cells that are marked by lowered levels of AMPK activity (and therefore more ATP), entosis provides the fittest cells in the population with large nutrient stores that support proliferation and population outgrowth.
Ferroptosis: necrotic cell death that spreads through cell populations.
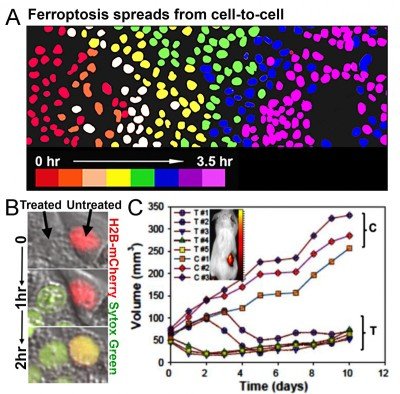
We have also recently discovered that depriving cells of essential amino acids and delivering specialized nanomaterials to the lysosome induces an iron-dependent form of cell death called ferroptosis (Fig. 3). Ultrasmall (6nm diameter) silica-based nanoparticles (PEG-C’ dots), which are in clinical trials for cancer detection studies at MSK, can bind and deliver iron from the extracellular environment into lysosomes. This leads to the induction of ferroptosis, which we found spreads through cell populations in a synchronous manner (Fig. 3). This form of cell death also has the ability to propagate from treated to untreated cells (Fig. 3), suggesting that ferroptosis could have potent anti-cancer activity. We indeed found that multi-dose intravenous particle delivery to xenograft tumor-bearing mice lead to tumor regression through ferroptosis. C’ dots are already in clinical trials for use as cancer detection agents, the result of a long-standing collaboration between Michelle Bradbury at MSK and Uli Wiesner, who developed the particles, at Cornell University. Our findings have opened up a new opportunity to use C’ dots as therapeutic agents to induce ferroptosis, which we are pursuing in pre-clinical studies in collaboration with Dr. Bradbury.
Nutrient Signaling and Scavenging Pathways
In addition to the regulation of cell death, the lab investigates mechanisms that regulate autophagy and lysosomes and how cells adapt to starvation by scavenging alternative sources of nutrients.
Autophagy Proteins Regulate Endocytic Mechanisms of Nutrient Scavenging
We and others have shown that the engulfment and degradation of bulk extracellular substrates, including whole cells engulfed by entosis or phagocytosis, and also macropinocytosed protein, can support proliferation when cells are deprived of essential nutrients such as amino acids. The macropinocytic ingestion of albumin from the extracellular environment in particular has been shown to promote the growth of Ras-mutant pancreatic cancers, and may be an important pathway that drives cancer progression in poorly vascularized tumors. Unlike autophagy that allows cells to recycle their own nutrients from intracellular substrates, these extracellular scavenging pathways allow starved cells to accumulate the biomass that is needed to support proliferation.
By examining the maturation of macropinosomes, phagosomes, and entotic vacuoles, we discovered that these extracellular scavenging pathways are cross-regulated by autophagy proteins. The autophagy protein microtubule-associated protein 1 Light Chain 3 (LC3) becomes lipidated onto macropinosomes, phagosomes, and entotic vacuole membranes in a manner requiring core autophagy genes (e.g., Atg5, Atg7) through a mechanism called non-canonical autophagy or LC3-Associated Phagocytosis (LAP) (Fig. 7). LC3 lipidation occurs on endocytic vesicles immediately prior to lysosome fusion and is required for the efficient degradation of engulfed substrates, suggesting that these scavenging pathways require machinery from the autophagy pathway for function. Our lab and others have now made important advances to elucidate key aspects of the regulation of autophagy proteins in this context, including identifying genetic features that distinguish LAP from canonical autophagy.
Regulation of Lysosomes and Nutrient Export by mTORC1 and PIKfyve
We have identified a late stage of macroendocytic vesicle maturation that is controlled by mTORC1, involving fission that shrinks the size of large lysosomal vesicles and redistributes engulfed, degraded cargo throughout lysosome networks. mTORC1 recruits specifically to lysosomes that harbor engulfed nutrients and controls their fission, suggesting that mTORC1 localizes to lysosomal membranes not only to sense amino acids but also to act as an important regulator of lysosome physiology. We have now also identified a second, parallel pathway that controls membrane fission, involving the lipid kinase PIKfyve and its effector calcium channel TRPML-1. Both mTORC1 and PIKfyve-TRPML1 participate in coordinating lysosomal membrane trafficking and the cytosolic export of nutrients but do not regulate the degradation of engulfed substrates, demonstrating complex regulation, post-lysosome fusion of macroendocytic vesicles. Our studies also identified PIKfyve as a potential drug target for pancreatic cancer, as we found that PIKfyve is required for the proliferation of cancer cells when nutrients are acquired through macropinocytic protein scavenging, an activity that is known to support pancreatic cancer growth.