Recombination, genomic integrity, and heterogeneity in B cells
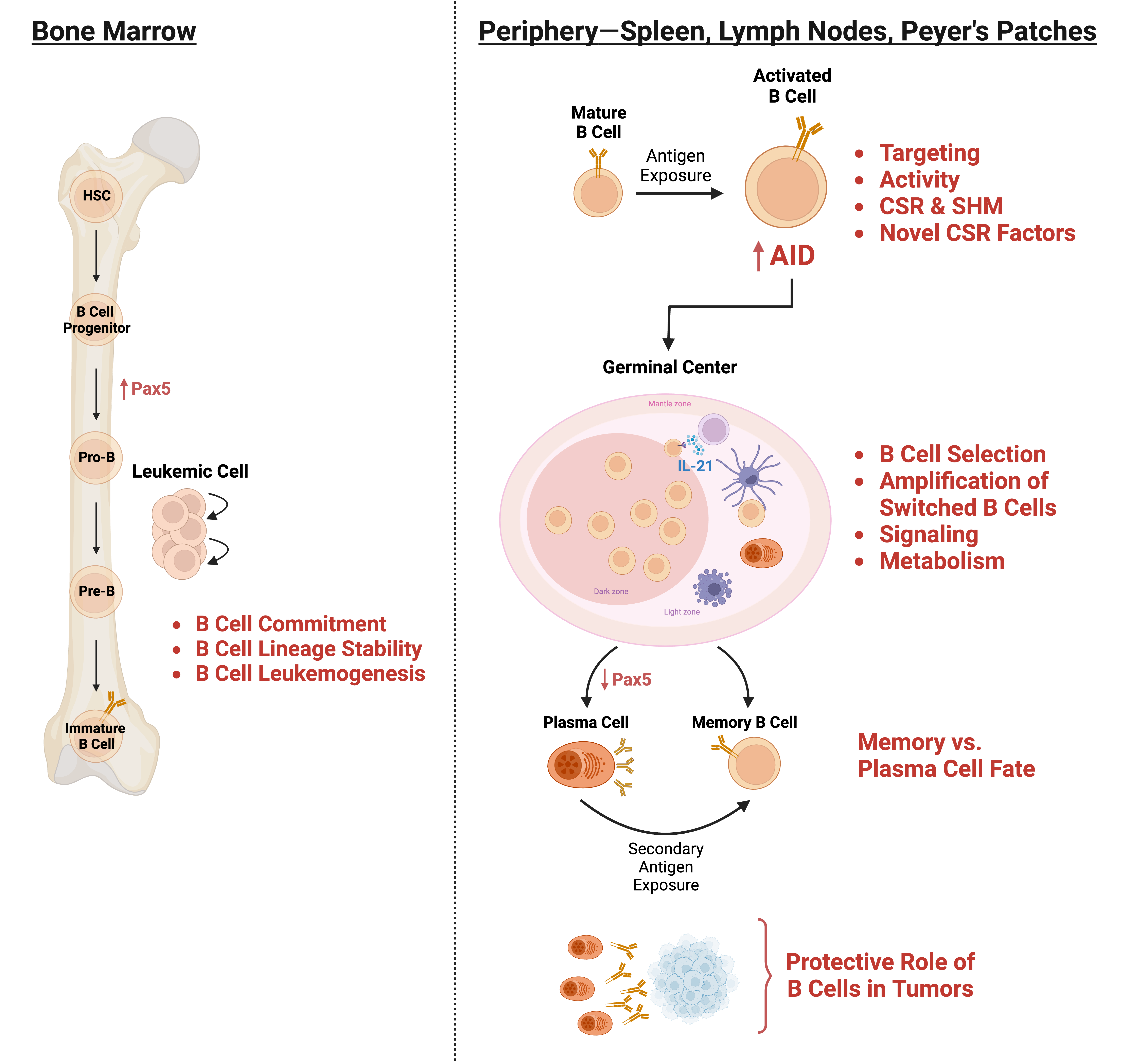
The focus of our lab is to decipher molecular cues that allow a mature B cell to undergo class switch recombination (CSR), a precisely tailored and finely tuned adaptive response essential for humoral immunity. CSR occurs through a DNA deletional-recombination event at the immunoglobulin heavy chain (IgH) locus so that the B cell changes from expressing IgM to one expressing a secondary antibody isotype such as IgG, IgE or IgA. A failure to undergo CSR leads to immunodeficiency syndromes. However, this otherwise beneficial and essential immune response has a dark side. CSR proceeds through the deliberate generation of DNA double strand breaks (DSBs), one of the most genome-destabilizing lesions that can occur in a cell. Indeed, mistargeted DSB formation outside the IgH locus and aberrant DNA repair are major drivers of B cell lymphomagenesis. Our overarching objective is to elucidate the mechanisms by which CSR is regulated to promote immunity while minimizing collateral damage associated with the process.
At the mechanistic level, CSR is an intricately choreographed interplay of transcription, DNA deamination, and DNA repair. The IgH locus comprises several constant region (CH) genes, each of which is preceded by transcribed, repetitive 1-12 kb switch (S) region DNA that serve as recombination targets. The B cell specific enzyme AID (for activation induced cytidine deaminase) deaminates cytidines to uridines at S regions to initiate a cascade of reactions ultimately leading to the generation of DSBs. End-joining of DSBs at donor and acceptor S regions (Sµ and Sε, respectively, in example shown in figure), primarily by the non-homologous end-joining (NHEJ) pathway, completes CSR. While work over the past couple of decades has provided an overall idea of this reaction, many key questions remain unanswered including the mechanisms by which AID activity is regulated and restricted to the IgH locus, the pathways that repair both IgH and non-Ig DSBs generated as a result of physiological and bystander AID activity, and processes that “brake” a primary immune response to maintain cellular heterogeneity. These unresolved issues constitute ongoing projects in the lab.
Non-coding RNA in recruitment of AID to DNA
The mechanism that specifically recruits AID to the IgH has baffled the field for a long time. We have now discovered a novel RNA-guided pathway that shepherds AID to switch regions (Zheng et al, Cell, 2015). We demonstrated that switch region RNA, generated at a post-splicing step, forms G-quadruplex structures, binds AID and recruits the deaminase to DNA. We are now engaged in examining the relevance of this RNA-guided pathway in both immunity and in sheltering non-IgH regions from AID-induced collateral damage. Additionally, RNA-mediated recruitment of AID to generate DSBs is reminiscent of CRISPR/Cas9-dependent DSB formation during an adaptive response in bacteria; ongoing experiments are geared to test if AID can be targeted to non-Ig regions in an RNA-dependent fashion. In addition to S region RNA in facilitating AID recruitment, we are actively investigating the role of microRNAs in regulating CSR (Pucella et al, Journal of Immunology, 2015).
Coupling the generation and repair of DSBs during CSR
Productive CSR requires end joining between donor and acceptor S regions that could be as far apart as 100 kb. An outstanding issue in CSR was how an otherwise robust NHEJ pathway is subverted from joining DSBs within individual S regions. We favored the notion that a high density of DSBs synchronously induced by AID could allow some to persist in the face of efficient repair to participate in CSR. We have now discovered that the DNA damage response protein ATM instigates a positive feedback loop through AID phosphorylation to couple induction of DNA damage with efficient DNA repair (Vuong et al, Nature Immunology, 2009; Vuong et al, Nature Immunology, 2013). Using a variety of mouse models and biochemical assays, we are engaged in elucidating the role of ATM in its canonical role in maintaining genomic integrity in B cells while simultaneously serving a counterintuitive function in promoting formation of DSBs.
Role of Holliday junction resolvases in repairing non-IgH DSBs
While NHEJ is the major pathway that repairs DSBs at S regions to complete CSR, we believe off-target DSBs are repaired through homologous recombination (HR). A critical but poorly understood aspect of HR is the “resolution” of Holliday junctions generated during the process. Two nucleases, Mus81 and Gen1, have been implicated at this resolution step. We have now generated mouse models in which one or both nucleases can be conditionally deleted in any cell, allowing us to examine their roles in B cell biology and in general DNA repair (in collaboration with the Jasin lab in the Developmental Biology Program at MSK).
Regulating B cell heterogeneity
Cellular heterogeneity is an essential feature of the adaptive immune system and is best exemplified during an immune response when an expanding B cell clone assumes multiple cell fates, including class-switched B cells, antibody-secreting plasma cells, and memory B cells. While each cell type is essential for immunity, their generation must be exquisitely controlled since a class-switched B cell cannot revert back to the parent isotype and a terminally differentiated plasma cell cannot contribute to the memory pool. Efficient generation of memory B cells, integral to successful vaccination and long-term immunity, would in principle require negative modulation of alternate cell fates. We have performed a screen to identify factors that could potentially regulate activation-induced cell-fate outcomes. Ongoing work aims to molecularly elucidate how these molecules orchestrate B cell heterogeneity.