The Lewis lab research interests are focused on the development of radiopharmaceuticals for targeted diagnosis and treatment of cancer. By combining small- and biomolecule-based targeting agents with positron-emitting or therapeutic radioisotopes, we can interrogate molecular profiles of cancer using noninvasive nuclear imaging or treat tumors specifically with endoradiotherapy. Our research program is a molecular-imaging-based program focused on the targeting of nonstandard nuclides, with an emphasis on developing these novel radiopharmaceuticals for clinical translation. A few examples of our work are given below.
The Lewis lab research interests are focused on the development of radiopharmaceuticals for targeted diagnosis and treatment of cancer. By combining small- and biomolecule-based targeting agents with positron-emitting or therapeutic radioisotopes, we can interrogate molecular profiles of cancer using noninvasive nuclear imaging or treat tumors specifically with endoradiotherapy. Our research program is a molecular-imaging-based program focused on the targeting of nonstandard nuclides, with an emphasis on developing these novel radiopharmaceuticals for clinical translation. A few examples of our work are given below.
Precision Imaging with 89Zr-based PET Agents
A primary research focus of our laboratory is the synthesis, development, and validation of Zr-89-based biomolecular imaging agents. One of the most fundamental principles in the construction of effective antibody-based nuclear imaging agents is matching the physical half-life of the radioisotope to the pharmacokinetic half-life of the targeting vector. This exigency has led our lab and others to seek to exploit the positron-emitting radiometal 89Zr (t1/2 = 78.41 h) as an effective and versatile long-lived radiolabel for PET imaging. Currently, our laboratory is pursuing a wide variety of projects that employ 89Zr as a radiolabel. The following examples, in particular, combine to paint a representative picture of the breadth of our work.
89Zr-5B1
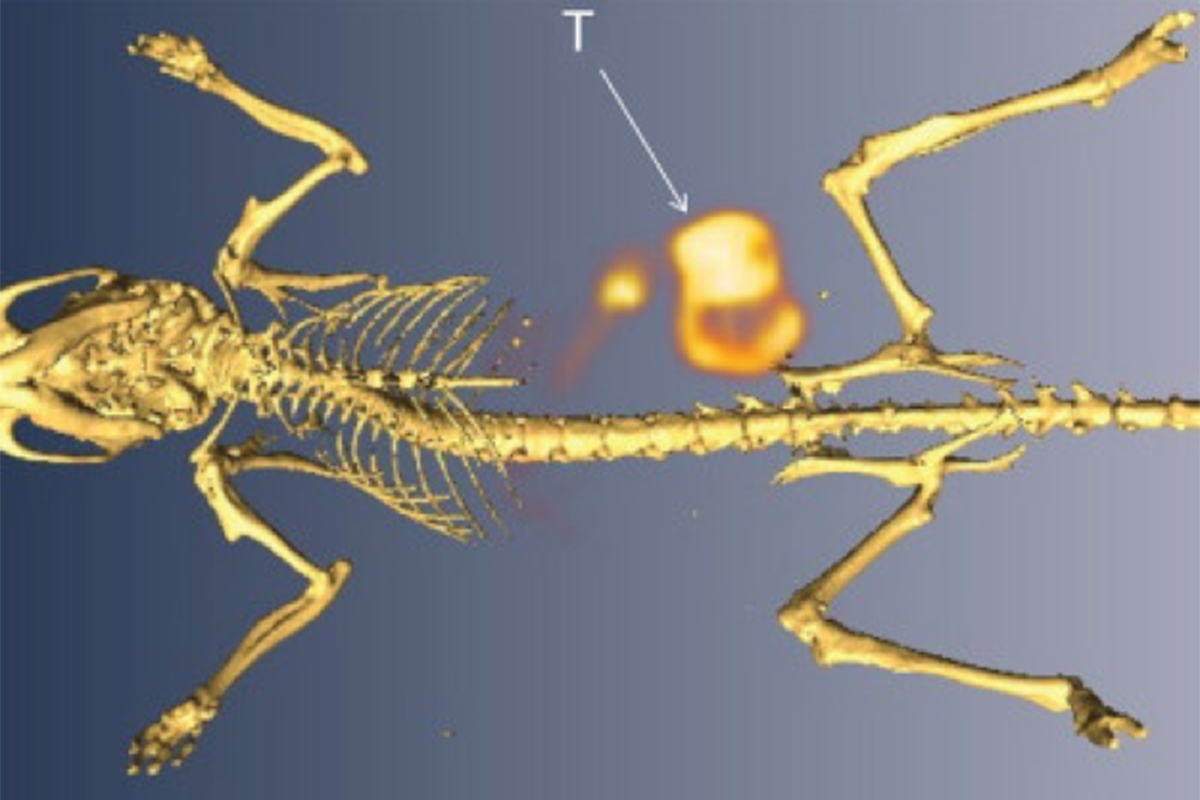
Serum circulating antigens continue to garner attention as molecular determinants of cancer. One of these antigens, CA19.9 or sialyl Lewis, has shown clinical value as a biomarker for diagnosis, stratification, and prognosis in pancreatic ductal adenocarcinoma (PDAC); currently, serological CA19.9 antigen assay is the standard clinical screening tool for detecting primary and metastatic disease and for evaluating response to therapy in PDAC. Other malignancies have also shown elevated levels CA19.9 in the blood, including those stemming from the colon, ovaries, and lung, which makes CA19.9 a target with broad utility across different cancer types. Despite these findings, serological assay is limited in its inability to identify the location and extent of malignancy, and furthermore, remains limited in its capacity for early detection of PDAC. However, observations of elevated tumor cell surface density of CA19.9 have enabled high-contrast molecular imaging of PDAC via PET and optical imaging.
Given the clinical potential of CA19.9, we have utilized a fully human, CA19.9-targeting monoclonal antibody, 5B1, as a tumor-targeting vector and radiolabeled it with 89Zr in collaboration with MabVax Therapeutics, Inc., the inventor of this antibody. Through PET imaging and biodistribution studies, we have demonstrated the exquisite specificity of 89Zr-5B1 for its target in murine models of PDAC (Fig. 1A). In later studies, we employed 5B1 for specific delivery of therapeutic radionuclides in mouse models. Our preclinical efforts have since culminated in translation of 89Zr-5B1 to the clinic in a first-in-man study.
89Zr-Transferrin
89Zr-transferrin offers a noninvasive technology for quantitatively measuring an oncogenic signaling pathway, more specifically the MYC status of tumors. 89Zr-transferrin binds the transferrin receptor 1 (TFRC, CD71) with high avidity, and we have shown this can be used to produce high-contrast PET images that quantitatively reflect treatment-induced changes in MYC-regulated TFRC expression in a MYC-driven prostate cancer xenograft model (Figure 2). Indeed, the agent has been shown to detect the in situ development of prostate cancer in a transgenic MYC prostate cancer model, as well as in prostatic intraepithelial neoplasia (PIN) before histological or anatomic evidence of invasive cancer. In view of these efforts and results, we are in the beginning stages of a Phase I trial for 89Zr-transferrin in prostate cancer patients. 89Zr-transferrin is also being pursued as an agent to improve the diagnosis of triple negative breast cancer as well as interrogate target engagement of small molecule therapies and assess oncogenic pathway intersection in lymphoma and pancreatic cancer.
Interrogating preclinical host biology using 89Zr-labeled antibodies
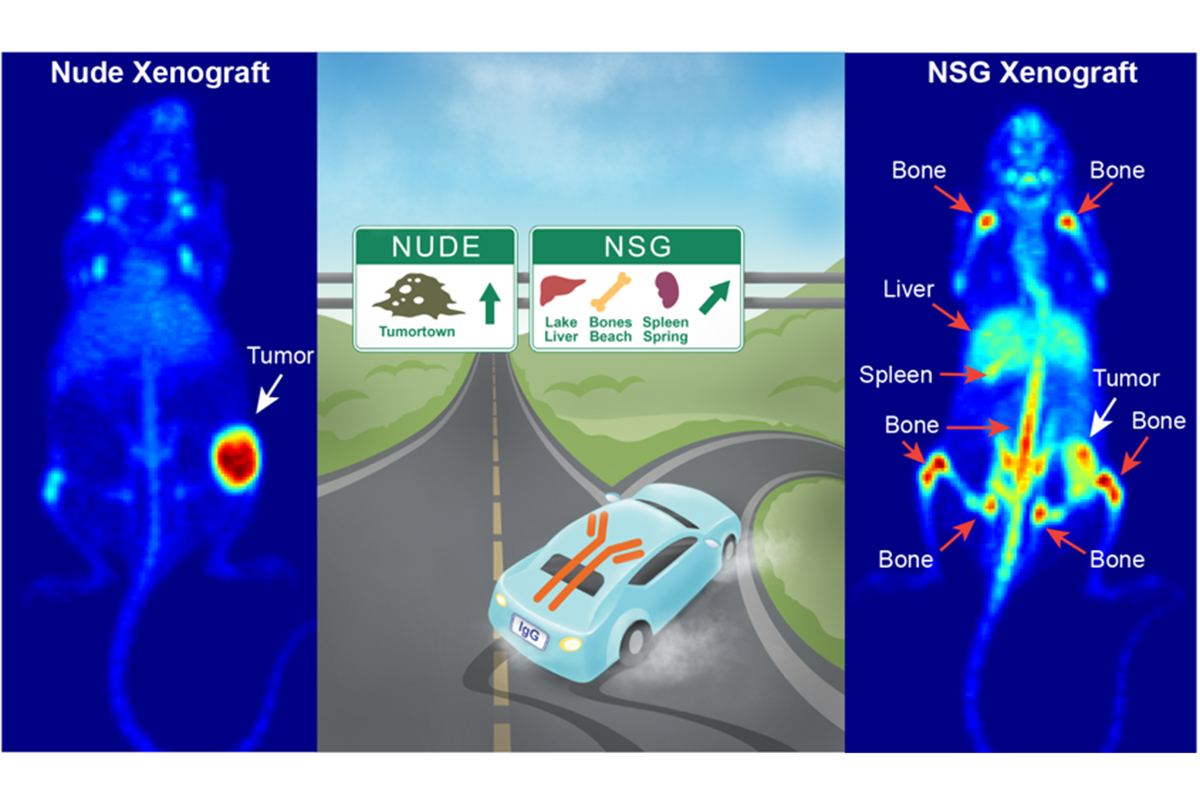
Translation of antibody-based therapeutics requires validation in preclinical models of human disease. Such models often necessarily rely on murine hosts engineered with immunodeficiency, and. Despite how often immune-deficient mouse models are used, the ultimate effect of these genetic modifications on the drug biodistribution remain relatively unexplored. We have demonstrated the impact of these effects on tumor targeting to be substantial, which highlights the need for consideration of host immunobiology in relation to antibody origin and glycosylation status in preclinical drug development (Figure 2).
Targeting the acidic microenvironment with pH (Low) Insertion Peptide (pHLIP)
The microenvironment of solid tumors is often characterized by a phenomenon known as the Warburg effect, an increased reliance on glycolytic respiratory pathways that results in the over-production of lactic acid. This excess lactic acid in turn requires extracellular transport to sustain tumor survival; however, the poorly formed vasculature of tumors hampers the subsequent diffusion of this acidity. As a result, the extracellular milieu in malignant tissues is relatively acidic (pHe~6.5–6.8) compared to that of normal cells (pHe~7.3-7.4). This reduced pH, often termed tumor acidosis, is an extremely promising target for noninvasive cancer imaging.
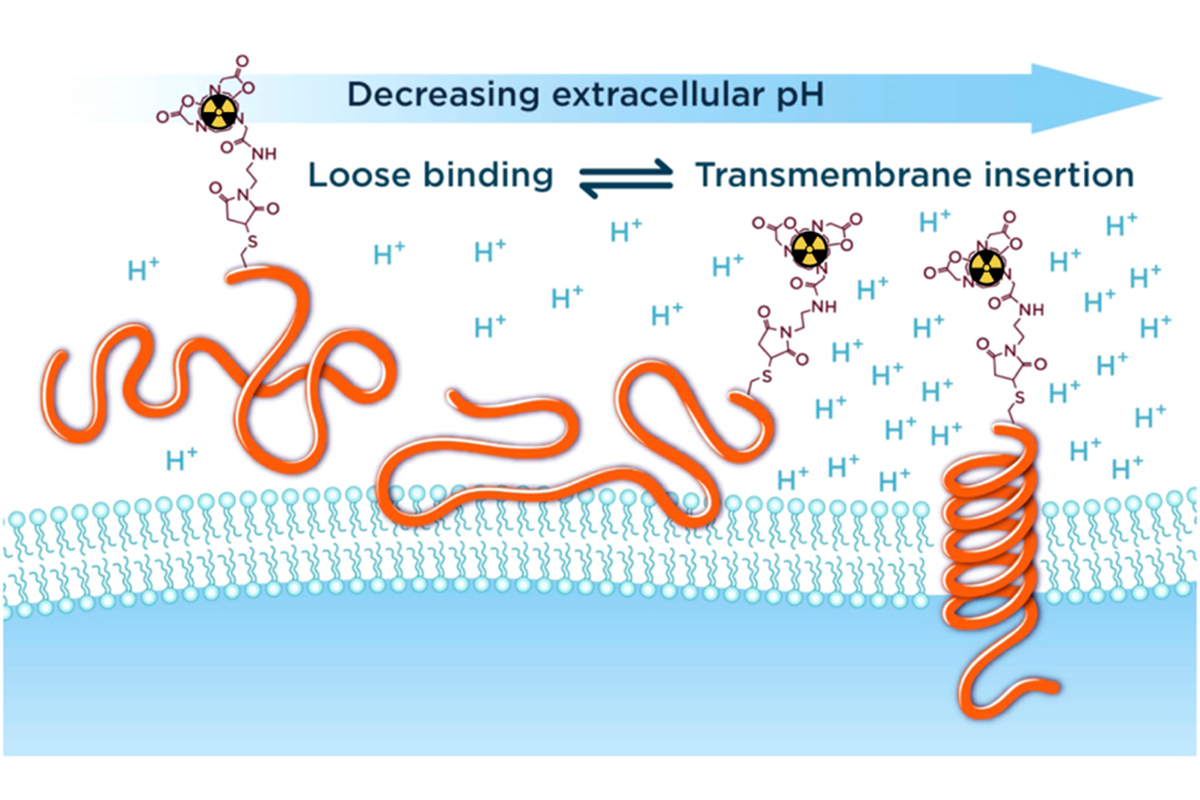
In partnership with Yana Reshetnyak and Oleg Andreev at the University of Rhode Island, Donald Engelman at Yale University, and pHLIP Technologies Inc., our laboratory is focused on developing a PET imaging agent for tumor acidosis based on a peptide carrier known as “pH (Low) Insertion Peptide” (pHLIP) (Figure 3). pHLIP is composed of a 22-37 amino acid sequence derived from the C-helix of Bacteriorhodopsin. As shown in Figure 3, in the acidic extracellular environment of a tumor, the peptide inserts in the cellular membrane, forming an α-helix and acting in essence as a nanosyringe. We have harnessed this peptide technology for imaging by conjugating radiometal chelates (68Ga, 64Cu) and 18F-fluorinated derivatives to various pHLIP variants and investigating the pharmacokinetic profile of these imaging agents in murine models of prostate cancer. The first generation pHLIP peptide is characterized by prolonged blood circulation, so in addition to working with this original variant, we have also continued to modify and improve the pharmacokinetics of pHLIP with various truncated sequences, radionuclide, and chelates.
Pretargeting cancer using bioorthogonal Diels-Alder click chemistry
The remarkable specificity and affinity of antibodies make them extremely attractive vectors for the delivery of diagnostic and therapeutic radioisotopes to cancer cells. Over the past two decades, a wide variety of antibody-based radiopharmaceuticals have been successfully developed, employing isotopes ranging from 89Zr for imaging to 225Ac for therapy. A significant limitation of the use of radiolabeled antibodies, however, is that their relatively slow pharmacokinetics mandate the use of therapeutic isotopes with multiday physical half-lives. This combination of long biological and physical half-lives gives rise to an important complication: high activity concentrations in and radiation doses to nontarget organs.
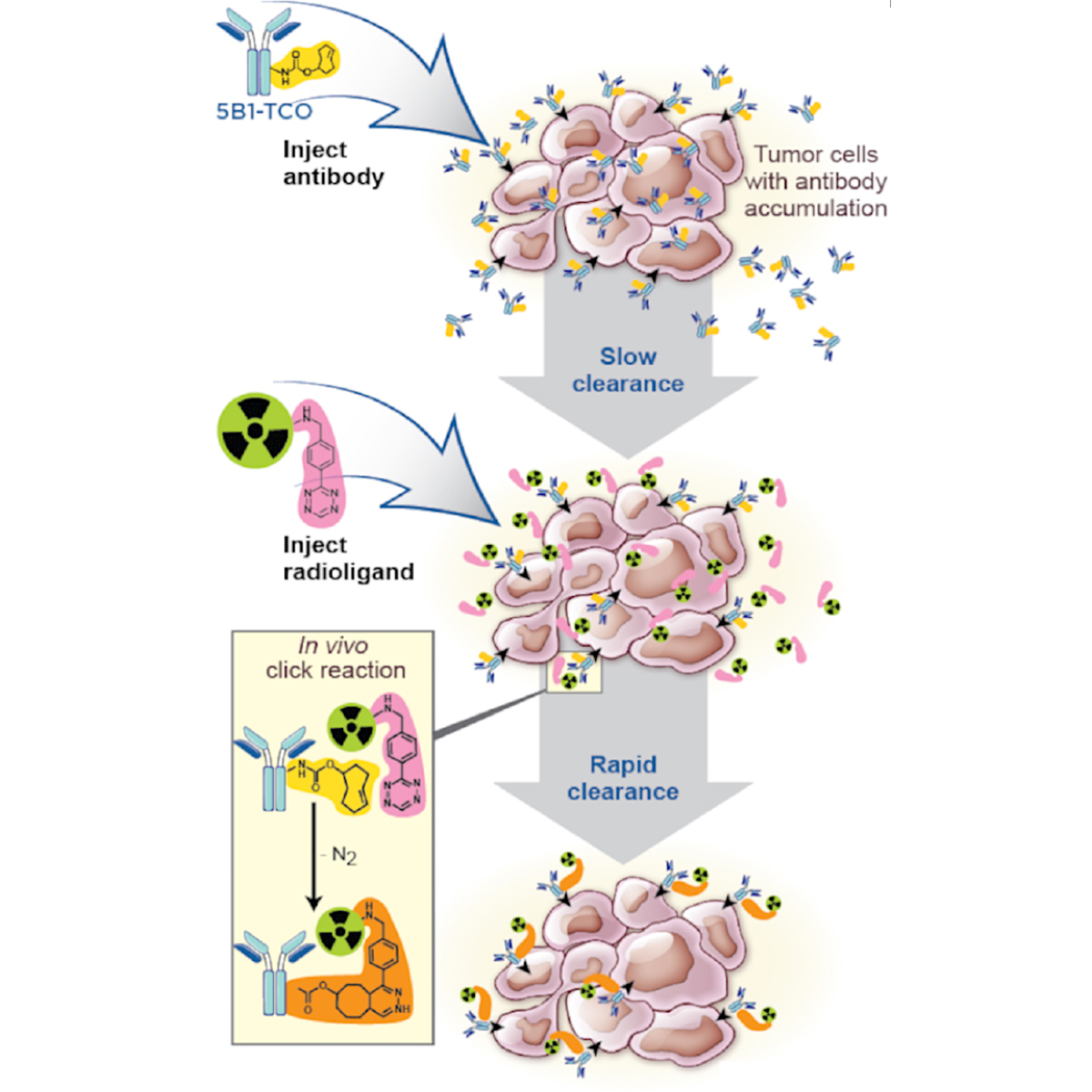
In order to circumvent this problem, considerable attention has been dedicated to the development of targeting methodologies that combine the advantages of antibodies with the pharmacokinetics of smaller molecules. One particularly appealing method (Figure 4) of achieving this balance while still employing intact antibodies is termed pretargeting. Generally, pretargeted methodologies involve four steps: (1) the injection into the bloodstream of a bivalent antibody with the ability to bind both an antigen and a radioligand; (2) the slow accumulation of the antibody in the tumor and concomitant clearance of the antibody from the blood; (3) the injection into the bloodstream of the small-molecule radioligand; and (4) the binding of the radioligand to the antibody followed by the rapid clearance of excess radioactivity.
In order to optimize pretargeting strategies, our lab has built and characterized an extensive library of tetrazine-based small molecules with a variety of pharmacokinetic profiles and a number of different chelating agents. The collection of chelating agents enables diversity in the choice of radioisotope, which can range from short-lived imaging isotopes (e.g. 68Ga, t1/2 = 68 m) to long-lived therapeutic isotopes (e.g. 225Ac, t1/2 = 10 d).
Currently, our laboratory is actively working on expanding the radiochemical applications of Diels-Alder click chemistry, with projects dedicated to the development of pretargeted radioimmunotherapy methodologies, the creation of pretargeted imaging strategies using peptide-based vectors, and the use of this biorthogonal click chemistry with nanoparticle-based theranostic agents.