The overall approach for this Lymphoma SPORE is to transform clinical practice by developing new treatments for diffuse large B cell lymphoma (DLBCL) and identify potential biomarkers that show how well these treatments are working.
This effort is organized into four projects:
Project Co-Leaders:
Hans-Guido Wendel (Basic)
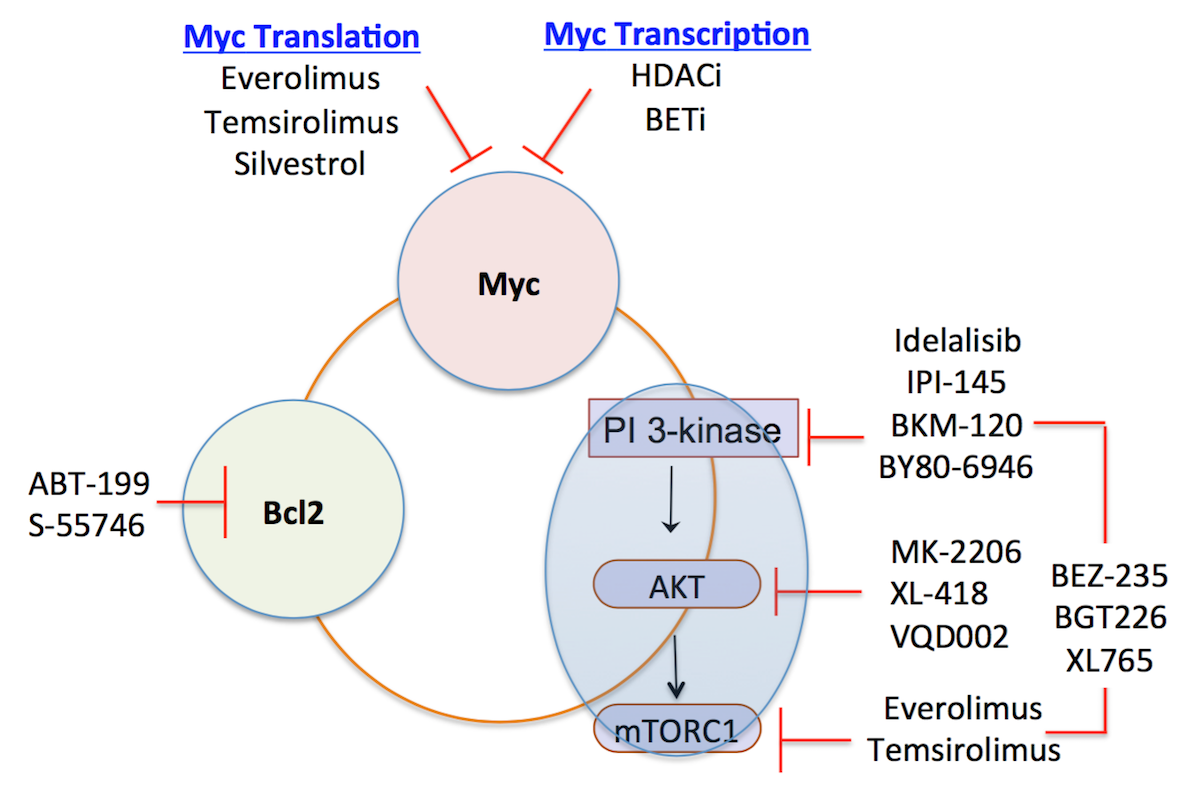
Recent studies demonstrated that ≈35% of DLBCL tumors co-express high levels of Myc and Bcl2 proteins (hereafter will be referred to as Myc+/Bcl2+ DLBCL). Except for the few cases involving Myc and Bcl2 translocations or genetic amplifications, mechanisms responsible for Myc and Bcl2 protein overexpression in DLBCL remain undetermined. Patients with Myc+/Bcl2+ DLBCL have a poor prognosis when treated with standard RCHOP regimen. Thus, Myc+/Bcl2+ DLBCL represents a biomarker-defined subset of patients with a clear unmet medical need. The major goal of this project is to develop novel, mechanism-based, treatment strategies to improve treatment outcome of patients with Myc+/Bcl2+ DLBCL. Based on preclinical data from our group and others, DLBCL cells frequently depend on several mechanisms to promote their growth and survival. Our focus will be on designing treatment strategies aimed at disrupting the oncogenic cooperation between Myc, and Bcl2, or Myc and activated PI3K signaling pathway (Fig 1). Our aims are:
Aim 1. To investigate the contribution of protein translation to Myc/Bcl2 expression in DLBCL.
Except for the few cases involving Myc and Bcl2 translocations or genetic amplifications, the mechanisms responsible for Myc and Bcl2 protein overexpression in DLBCL remain undetermined. Understanding the mechanism responsible for Myc and Bcl2 expression may provide opportunities for novel treatment strategies.
Aim 2. To develop mechanism-based treatment strategies for Myc+/Bcl2+ DLBCL
- Targeting Myc and Bcl2 oncogenic cooperation in DLBCL
- Targeting Myc and PI3K/AKT/mTOR pathway oncogenic cooperation in DLBCL
Aim 3. To evaluate the safety and efficacy of novel therapies developed and prioritized from Aims 1 and 2 in phase I/II studies in patients with Myc+/Bcl2+ DLBCL
Project Leader:
Renier Brentjens
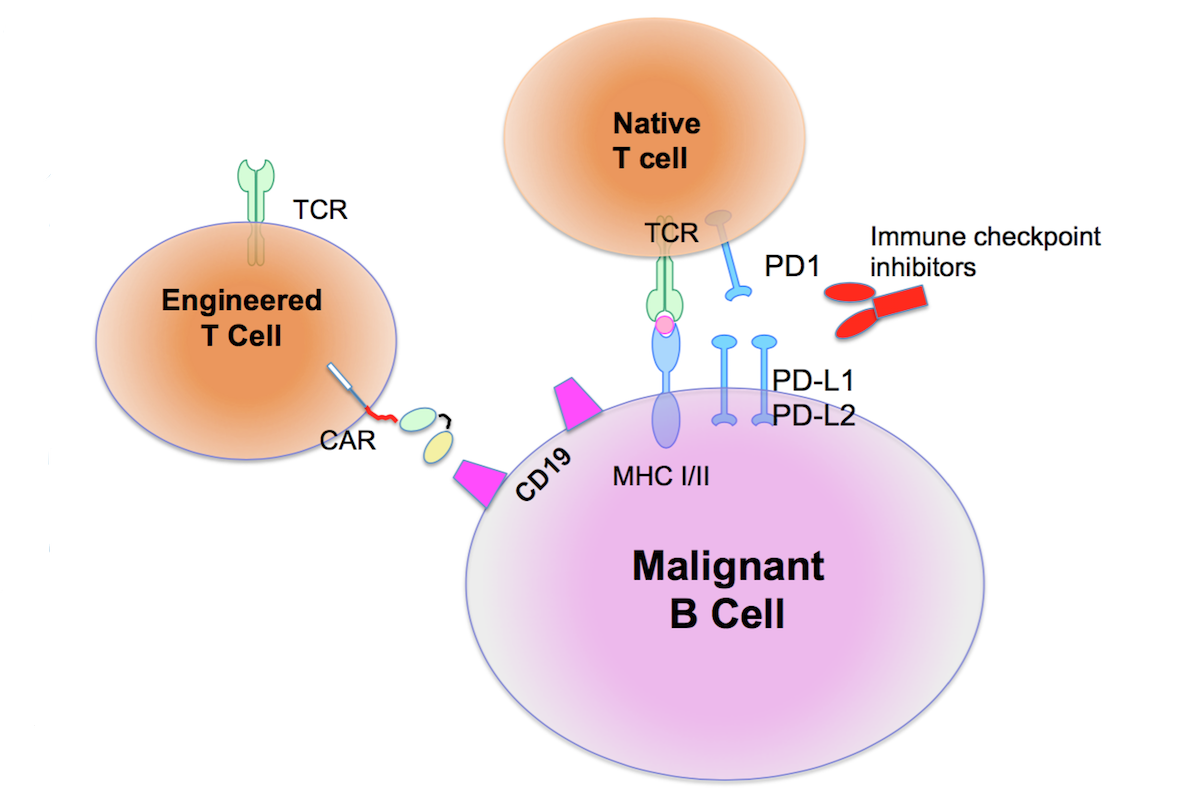
Despite currently available chemotherapy regimens, patients with relapsed DLBCL ineligible for autologous stem cell transplant (ASCT) have a very poor prognosis and represent an area of unmet need. One novel approach is the adoptive transfer of T cells genetically modified to express artificial T cell receptors termed CARs designed to recognize target antigens expressed on the tumor cell surface. A patient’s own T cells may be isolated and modified to express the CAR thereby redirecting T cell specificity to the tumor associated antigen. Most B cell non-Hodgkins lymphomas (NHL), including DLBCL, express the B cell specific antigen CD19. We and several other groups have recently reported initial clinical outcomes of patients with both low grade as well as aggressive B cell cancers treated with CAR T cells targeted to the CD19 antigen. To date, these clinical studies have reported marked anti-tumor responses in a variety of B cell malignancies especially in patients with low grade B cell CLL as well as even more impressive clinical outcomes in patients with relapsed B-ALL. However, there is a limited data on patients with aggressive DLBCL treated with CD19 targeted CAR T cells. An additional and relevant immune-based approach to cancer therapy not yet fully explored in the setting of B cell malignancies in general, and specifically in the setting of DLBCL is immune-checkpoint blockade through infusion of antagonistic monoclonal antibodies (MAb’s) targeted to the T cell PD-1 receptor, which when engaged with either the PD-L1 or PD-L2 ligand induces T cell anergy. As a result, blockade of this T cell checkpoint pathway with PD-1 specific MAbs in turn enhances the anti-tumor function of tumor targeted T cells. The goal of this project is to initially apply our CD19 targeted CAR T cell approach to relapsed ASCT ineligible DLBCL patients in a clinical trial as a single agent therapy following salvage chemotherapy. We will next investigate the rationale of combining 19-28z CAR T cell therapy with PD-1 checkpoint inhibition wherein the latter immune-based approach may protect the CD19 targeted CAR T cells from PD-L1 and PD-L2 mediated anergy. We will initially study this hypothesis in a clinically relevant syngeneic immune competent model of disease with subsequent translation in a planned second clinical trial targeting the same patient population. The specific aims of Project 2 are:
Aim 1. To conduct a Phase I/II clinical trial treating elderly transplant ineligible patients with relapsed DLBCL with salvage chemotherapy followed by CD19 targeted 19-28z CAR modified autologous T cells.
Aim 2. To optimize the efficacy of CAR modified T cell therapy through additional PD-1 checkpoint blockade.
Aim 3. To conduct a Phase I/II clinical trial treating elderly transplant ineligible relapsed DLBCL patients with salvage chemotherapy followed by CD19 targeted CAR T cells in combination with PD-1 blocking MAb.
Project Co-Leaders:
John Leonard (Clinical)
Ari Melnick (Translational/Basic)
DLBCL are subjected to substantial levels of stress. Sources of stress for these cells include massive proliferation, genomic instability, starvation, hypoxia, and suppression of checkpoint proteins that normally maintain homeostasis. Stress pathway activation enables B-cells to survive long enough for sub-population of B-cells to emerge acquiring many genetic mutations that facilitate malignant transformation. Our data indicate that DLBCLs are often addicted to a stress protein called tumor enriched Hsp90 (TE-Hsp90), which is essential for the actions of multiple oncoproteins. Along these lines the Chiosis lab developed PUH71, an inhibitor with selective activity against TE-Hsp90. In pre-clinical studies PUH71 was shown to potently kill DLBCLs without toxicity to other organs. PUH71 preferentially accumulates in tumors but not in normal tissues in animals and in humans. Indeed PUH71 distribution and tissue concentration can be precisely measured in humans using a novel PET scan method. PUH71 phase 0 and phase I studies are nearing completion, and have proven that PUH71 PET imaging provides an accurate measurement of tumor drug concentration and exposure (area under the curve), which in turn correlates with tumor responsiveness. Hence PUH71 is a potentially unique personalized companion biomarker for drug activity that may allow treatment to be individually tailored to patients. Herein we propose to perform the first phase II trial of PUH71, in patients with relapsed refractory DLBCL, accompanied by PUH71 PET non-invasive imaging, as well as correlative science to evaluate putative biomarkers that have emerged from pre-clinical laboratory studies.
Aim 1. Perform a PUH71 phase II clinical trial in patients with DLBCL
Aim 2. Compare and contrast spectrum of activity among DLBCLs to Hsp90 and Hsp70 inhibitors.
Aim 3. Build rational combinatorial regimens anchored on PUH71 and Hsp70 inhibitors.
Project Co-Leaders:
Riccardo Dalla-Favera (Basic)
Andrew Zelenetz (Clinical)
DLBCL represents the most common form of B-cell non-Hodgkin Lymphoma (B-NHL), accounting for ~30% of the de-novo diagnoses54. A significant fraction of DLBCL remains incurable, underscoring the need to identify molecular mechanisms that are responsible for disease development and that can be targeted therapeutically. Toward this end, we have integrated next-generation whole-exome sequencing analysis and high-density single nucleotide polymorphism (SNP) array analysis for the genome-wide detection of mutations and gene copy number changes in DLBCL. This combined approach has shown that DLBCL is frequently associated with structural alterations inactivating CREBBP (CBP) and, more rarely, EP300 (p300), two highly related histone and non-histone acetyltransferases (HATs) that act as transcriptional co-activators in multiple signaling pathways. Overall, ~37% of DLBCL and 36% of follicular lymphoma (FL) cases display genomic deletions and/or somatic point mutations that remove or inactivate the HAT-coding domain of these two genes. Moreover, 64% (n=25/39) of DLBCL derived from FL transformation harbor genetic lesions affecting these two loci. Notably, CBP lesions appear to represent early events acquired during the clonal expansion of a common mutated precursor cell, suggesting a role in the initial phases of tumorigenesis. We have demonstrated that CBP/p300 inactivation leads to specific defects in the acetylation-mediated inactivation of the BCL6 onco-protein and activation of the p53 tumor suppressor, therefore suggesting one possible mechanism by which these lesions may contribute to cell transformation. Based on these results, the general objectives of this project are to elucidate the normal and pathologic role of CBP and p300 in mature B cells, to establish pre-clinical models for their therapeutic targeting, and to assess the efficacy of drugs targeting deacetylation mechanisms in clinical trials. To achieve these objectives, the following Specific Aims will be pursued:
Aim 1. Identify the full complement of genetic and epigenetic lesions affecting CBP/p300 and investigate their pathogenetic and clinical impact in DLBCL.
Aim 2. Identify biomarkers of CBP/p300 activity in GC B cells that are deregulated in patients harboring CBP/p300 functional loss
Aim 3. Elucidate the role of CBP inactivation in lymphomagenesis in vivo.
Aim 4. Evaluate the safety and efficacy of novel therapies developed from Aims 1 and 2 in patients with relapsed DLBCL