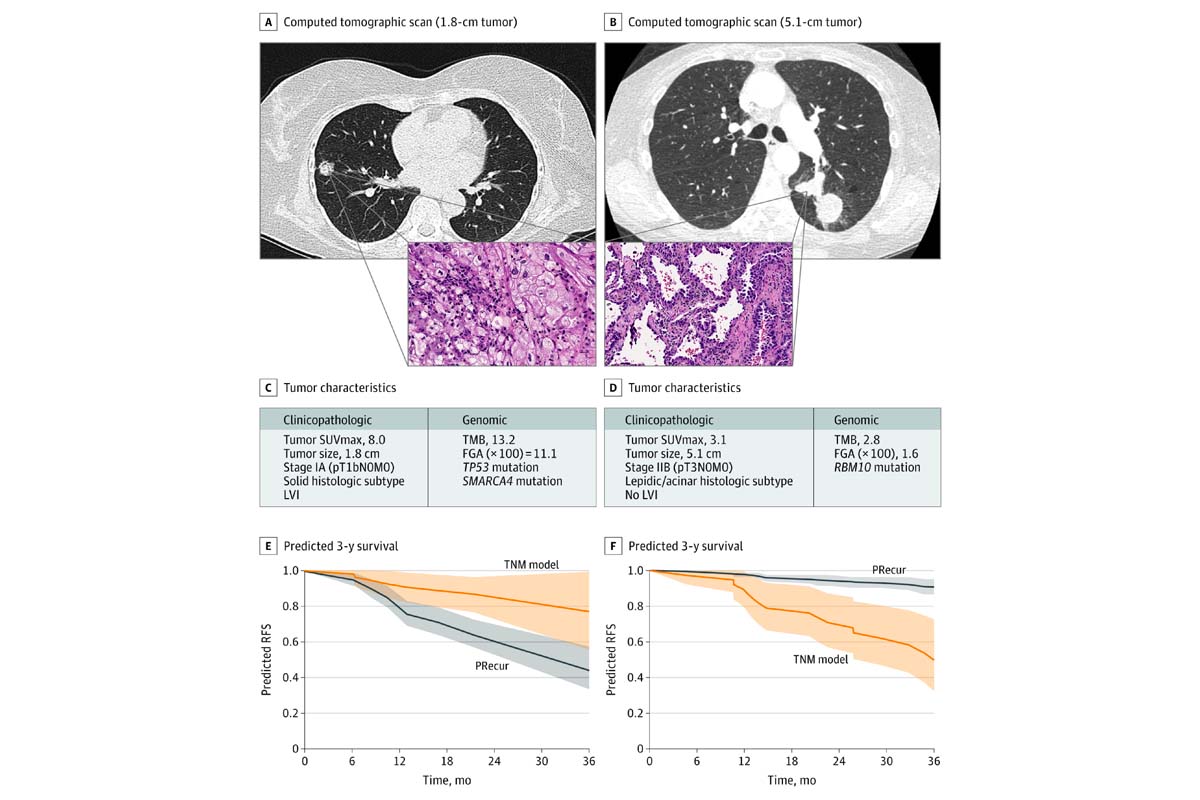
Despite complete surgical resection of stage I-III lung cancer, tumor recurrence develops in up to 50% of patients. Following recurrence, survival is poor, with five-year survival rates as low as 30% even with adjuvant cytotoxic chemotherapy for high-risk populations. The current rationale for recommending adjuvant therapy relies solely on TNM (tumor, node, and metastasis) classification, and more recently PD-L1 expression, and is largely agnostic to clinicopathologic and tumor genomic factors. As a result, survival benefits are modest at best when using the TNM model to select patients for adjuvant therapy. With the recent explosion of next-generation sequencing, our lab is rigorously investigating associations between tumor genomics, recurrence, and disease-free survival (DFS) in patients with early-stage lung adenocarcinoma (LUAD). In addition to a gene-centric approach, we have performed oncogenic pathway analyses to ascertain if these analyses can supplement existing TNM indicators for prognosis and prediction of recurrence. Using our large genomically annotated LUAD database, we were able to demonstrate unique differences in tumor genomics and mutational signatures across LUAD histologic subtypes. We have also identified the KRAS G12C mutation as a significant negative prognostic variable following complete resection of early stage LUAD4. We are interested in the prognostic abilities of tumor mutation burden (TMB), as well as the fraction of genome (FGA) - a measure of the percentage of the genome affected by gains or losses in copy number - and their association with LUAD recurrence. We developed PRecur, a novel computational machine-learning model that integrates genomic and clinicopathologic factors, to predict DFS in our MSK LUAD cohort (see figure below) and found that incorporation of both genomic and selected high-risk clinicopathologic variables is superior to TNM classification in predicting recurrence following surgery. We believe that continued investigation of tumor genomics in early-stage lung cancer has the possibility of moving the field in the direction of more personalized therapeutic decision making. Most recently, we analyzed thousands of LUAD samples to identify clinicopathologic and genomic features associated with metastasis, metastatic burden, organotropism, and metastasis-free survival. We found that inactivating mutations of TP53, SMARCA4, and CDKN2A correlate with a site-specific shorter time to metastasis. We actively collaborate with the Marie-Josée and Henry R. Kravis Center for Molecular Oncology at MSK, the Nikoulaus Schultz Laboratory, the Marcin Imielinski Laboratory, as well use MSK OncoKB®, MSK-IMPACT™, and related bioinformatics platforms.

- Brandt W, Bouadallah I, Tan KS, Park BJ, Adusumilli PS, Molena D, Bains MS, Huang J, Isbell JM, Bott M, Jones DR. Predictors of distant recurrence following R0 lobectomy for pN0 lung adenocarcinoma. J Thorac Cardiovasc Surg 2018; 155:1212-24.
- Zhou J, Sanchez-Vega F, Tan KS, Brandt WS, Jones GD, Caso R, Yan S, Adusumilli PS, Bott MJ, Huang J, Isbell JM, Sihag s, Molena D, Rusch VR, Chatila WK, Rekhtman N, Ladanyi M, Solit DB, Berger MF, Schultz N, Jones DR. Oncogenic pathway alterations are associated with disease-free survival in surgically resected lung adenocarcinoma. Clin Cancer Res 2019;25:7475-84.
- Caso R, Sanchez-Vega F, Tan KS, Mastrogiacomo B, Zhou J, Jones GD, Nguyen B, Schultz N, Connolly JG, Brandt WS, Bott MJ, Rocco G, Molena D, Isbell JM, Liu Y, Mayo MW, Adusumilli PS, Travis WD, Jones DR. The underlying tumor genomics of predominant histologic subtypes in lung adenocarcinoma. J Thorac Oncol 2020;15:1844-56.
- Jones GD, Brandt WS, Shen R, Sanchez-Vega F, Tan KS, Martin A, Zhou J, Berger M, Solit DS, Rizvi H, Liu Y, Adamski A, Chaft J, Riely G, Rocco G, Bott MJ, Travis WD, Rekhtman N, Molena D, Adusumilli PS, Imielinski M, Li B, Jones DR. A genomic-pathologic annotated risk model to predict recurrence in early-stage lung adenocarcinoma. JAMA Surg 2021;156(2):e205601.
- Jones GD, Caso R, Tan KS, Mastrogiacomo B, Sanchez-Vega F, Liu Y, Connolly JG, Murciano-Goroff Y, Bott MJ, Adusumilli PS, Molena D, Rocco G, Rusch VW, Misale S, Yaeger R, Drilon A, Arbour KC, Riely GJ, Rosen N, Lito P, Zhang H, Lyden DC, Rudin CM, Jones DR, Li BT, Isbell JM. KRASG12C mutation is associated with increased risk of recurrence in surgically resected lung adenocarcinoma. Clin Cancer Res 2021;27:2604-12.
- Lengel HB, Connolly JG, Jones GD, Caso R, Zhou J, Sanchez-Vega F, Mastrogiacomo B, Isbell JM, Li BT, Liu Y, Rekhtman N, Jones DR. The emerging importance of tumor genomics in operable non-small cell lung cancer. Cancers 2021;13(15):3656.
- Nguyen B, Fong C, Luthra A, Smith SA, DiNatale RG, Nandakumar S, Walch H, Chatila WK, Madupuri R, Kundra R, Bielski CM, Mastrogiacomo B, Donoghue MTA, Boire A, Chandarlapaty S, Ganesh K, Harding JJ, Iacobuzio-Donahue CA, Razavi P, Reznik E, Rudin CM, Zamarin D, Abida W, Abou-Alfa GK, Aghajanian C, Cercek A, Chi P, Feldman D, Ho AL, Iyer G, Janjigian YY, Morris M, Motzer RJ, O’Reilly EM, Postow MA, Raj NP, Riely GJ, Robson ME, Rosenberg JE, Safonov A, Shoushtari AN, Tap W, Teo MY, Varghese AM, Voss M, Yaeger R, Zauderer MG, Abu-Rustum N, Garcia-Aguilar J, Bochner B, Hakimi A, Jarnagin WR, Jones DR, Molena D, Morris L, Rios-Doria E, Russo P, Singer S, Strong VE, Chakravarty D, Ellenson LH, Gopalan A, Reis-Filho JS, Weigelt B, Ladanyi M, Gonen M, Shah SP, Massague J, Gao J, Zehir A, Berger MF, Solit DB, Bakhoum SF, Sanchez-Vega F, Schultz N. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell. 2022;185(3):563-575.e11.
- Lengel HB, Mastrogiacomo B, Connolly JG, Tan KS, Liu Y, Fick CN, Dunne EG, He D, Lankadasari MB, Satravada BA, Sun Y, Kundra R, Fong C, Smith S, Riely GF, Rudin CM, Gomez DR, Solit DB, Berger MF, Li BT, Mayo MW, Matei I, Lyden DC, Adusumilli PS, Schultz N, Sanchez-Vega F, Jones DR. Genomic mapping of metastatic organotropism in lung adenocarcinoma. Cancer Cell 2023;41(5):970-985.e3.