One of the major activities of the Geoffrey Beene Cancer Research Center is to provide investigators with seed funding for new and innovative projects. Below are some examples of previous and current grant recipients who have made major, cutting-edge advances in cancer research.
Grant recipient in 2013-2015
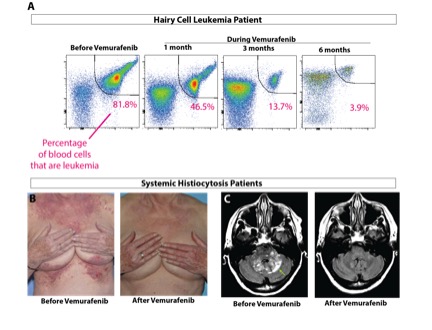
Omar Abdel-Wahab has received grants from the Geoffrey Beene Cancer Research Center to study two forms of rare blood cancer driven by mutations in a protein called BRAF: hairy cell leukemia and systemic histiocytosis. Although BRAF mutations are quite common in solid tumors such as melanoma and colon cancer, they are almost never seen in blood cancers except for these two poorly understood diseases. Dr. Abdel-Wahab and his colleagues were particularly interested in these diseases because the BRAF inhibitor vemurafenib (Zelboraf®) had proven very effective for patients with BRAF-mutant melanoma. They therefore carried out two separate clinical trials for each condition, both of which were recently published in the New England Journal of Medicine. In both studies, they found that patients with hairy cell leukemia or histiocytosis who had disease refractory to conventional chemotherapy responded exceptionally well to vemurafenib. These results are now being used to seek FDA approval for vemurafenib in these disorders.
While doing this work, Dr. Abdel-Wahab and his team learned that 100 percent of patients with hairy cell leukemia have the BRAF mutation, while only 50 percent of histiocytosis patients have it. This led to the discovery that the other half of histiocytosis patients have mutations in other members of the BRAF signaling pathway and respond to other types of targeted therapies. They are now initiating a clinical trial of one these new drugs, called cobimetinib (Cotellic™), for histiocytosis patients that lack the BRAF mutation. The results of this treatment in the first few patients have been extremely encouraging.
Grant recipient in 2011-2013
The concept of precision cancer medicine is based on the premise that each patient’s cancer is unique and should be treated based on the specific mutations and molecular changes in the tumor. A growing number of cancer drugs target cancers with particular genetic mutations, but in order to provide effective therapies to more patients, we must know more about each patient’s tumor individually. High-throughput mutation profiling of the DNA from every cancer for the mutational status of every known cancer gene is central for the success of precision cancer medicine.

With the support of the Geoffrey Beene Cancer Research Center, Michael Berger’s lab developed a test called MSK-IMPACT™, which is capable of detecting all possible mutations in 341 genes with the greatest clinical or biological significance for cancer. Through extensive experimentation, Dr. Berger and colleagues optimized the performance of the test for a variety of clinical specimens. Following initial success in pilot studies, Dr. Berger has since deployed production-scale MSK-IMPACT testing retrospectively for translational research and prospectively for clinical diagnosis of patients.
In the last three years, Dr. Berger and his colleagues have published more than 40 research articles describing studies with MSK-IMPACT to identify clinically relevant mutations. Most significantly, the MSK-IMPACT platform has been successfully implemented in Memorial Sloan Kettering’s clinical molecular diagnostics laboratory to directly inform treatment decisions. Based largely on the success of the work funded by the Geoffrey Beene grant, Dr. Berger assumed oversight of the validation of MSK-IMPACT as a clinical test. He obtained full approval from the New York State Department of Health allowing him to report results back to patients and their doctors. This promises to improve treatment decisions for numerous patients and to preidentify patients eligible for current and future clinical trials. As of January 2016, more than 8,000 MSK patients had received MSK-IMPACT testing on their tumors.
Grant recipient in 2013-2015


Over the past two years, the Chen laboratory has established 15 such “organoid” lines from patients treated at Memorial Sloan Kettering. Each line harbors a different set of mutations; together, they encompass a large repertoire of the genetic diversity found in prostate cancer. The researchers are now using these lines to understand how mutations affect the clinical presentation of the disease and the sensitivity of the tumor to different drugs or drug combinations. These new models should greatly accelerate the pace at which new treatments can be delivered to prostate cancer patients.
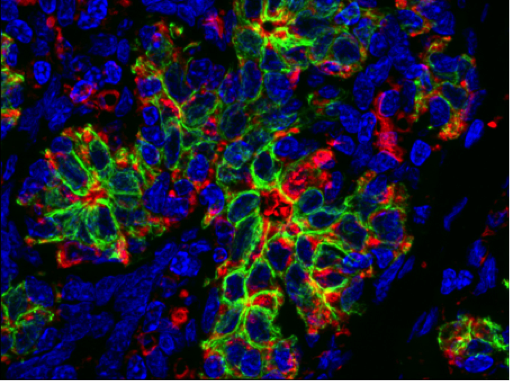
Most cancer deaths occur because the disease spreads, or metastasizes, from the original tumor site to a distant location in the body. In breast cancer patients, metastasis to the brain can occur months or years after seemingly successful treatment of the primary tumor. Johanna Joyce, an inaugural Geoffrey Beene Jr. Chair recipient, and colleagues have found that a protein called cathepsin S may play a key role in the spread of breast cancer to the brain. A complex interplay between breast cancer cells and certain surrounding cells called macrophages induces both cell types to secrete increased levels of cathepsin S, an enzyme that promotes the cancer cells’ ability to metastasize. Dr. Joyce’s lab recently showed that cathepsin S could be an important target for new drugs. The Joyce lab was interested in how these noncancerous cells at metastatic sites react when they encounter the tumor cells. To investigate these questions, the research team studied mice implanted with human breast cancer cell lines that are known to spread to the brain, bones, and lungs. They looked at how both the tumor and its surrounding microenvironment change during cancer spreading. The cancer cells and surrounding cells were subjected to genetic analysis to see which genes were more active than normal. In mice with brain metastases, the cathepsin S gene was significantly overexpressed. Interestingly, the tumor cells produced more cathepsin S at the beginning of metastasis and then tapered production, in parallel with a progressive increase in macrophage cathepsin S levels within the brain.
The researchers showed that cathepsin S enables the cancer cells to penetrate through the blood-brain barrier, a densely packed vascular structure that protects the brain from most large molecules in the blood. They found that cathepsin S cleaves a protein called (JAM)-B, which normally helps hold cells in the blood-brain barrier together. The higher levels of cathepsin S presumably negate (JAM)-B’s effect and allow the cancer cells to penetrate the barrier.
To validate the role of cathepsin S in promoting brain metastases, the researchers inhibited it with experimental drugs. This significantly reduced the development of brain metastasis in the animals. The Joyce lab would like to move forward with clinical trials for cathepsin S inhibitors in the near future.

With support from the Geoffrey Beene Cancer Research Center, Ross Levine’s group demonstrated that these mutations influence the aggressiveness of AML. They then used these data to identify clinical implications of genetic alterations in AML, and to develop a clinically useful way to predict overall outcome in AML, improving the ability of oncologists to decide on the best treatment strategy. As one example, their efforts identified certain subsets of AML patients that benefit from dose-intensified chemotherapy, which allows oncologists to spare patients who are unlikely to benefit from this more toxic regimen. This work has led to the widespread use of mutational profiling of AML patients and to the development of state-of-the-art genomic profiling assays for patients at MSK and elsewhere.
Awarded 2019: Imaging and therapy of neuroendocrine prostate cancer using DLL3-targeting antibodies

Colorectal cancer is the third most common cancer in developed countries. Despite improvements in screening and early detection, it also remains the second overall cause of cancer-related death. A goal of Scott Lowe’s laboratory is to understand what genes are responsible for causing colon cancer to grow and spread in humans. This information is essential for developing new, personalized, targeted therapies to cure the disease.
Although many genes have been implicated as drivers of colon cancer, perhaps the most important is a gene called APC. One function of APC is to control cell division in the colon. After cells in the colon have performed their job of absorbing nutrients, water, and minerals from ingested food, they are shed and replaced by new ones. This process is dependent on APC, and mutations in this gene interrupt the process. As a result, colon cancer cells are not shed and instead remain behind and grow in the colon as a tumor. If left untreated, this tumor can eventually gain additional mutations, causing it to invade surrounding tissues and distant organs.
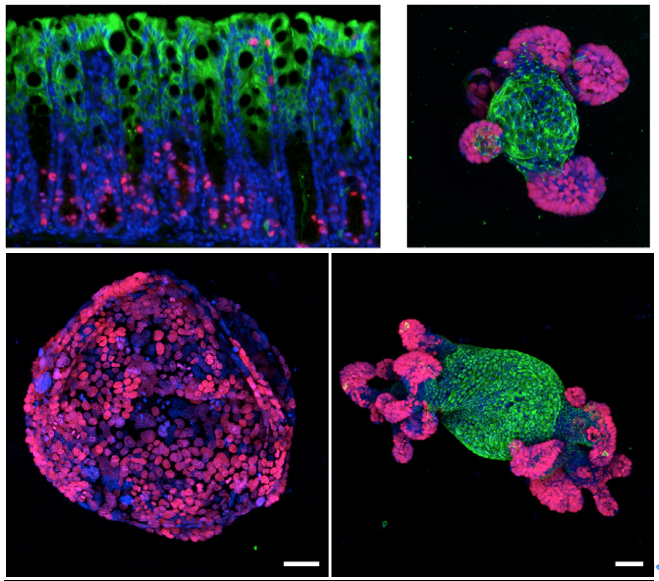
With support from the Geoffrey Beene Cancer Research Center, the Lowe Lab has developed a unique system in which APC function can be turned off and on. They showed that when they turned off APC function, mice developed colon tumors that very closely mirrored human colon cancer. More importantly, when they restored APC in advanced tumors that were invasive, the cancer was completely eliminated and the mice were cured of their disease.
They confirmed these findings by studying colon cancer cells growing in tissue culture plates. Normal intestinal cells form organoids, or “mini-guts,” on these plates, recreating the process of cell division, cell shedding, and replacement by new cells. When they disrupted APC gene function in these mini-guts, this process stopped and the cells grew as a single large sphere, much like tumors in the mice. However, just like the mice, when they restored APC function, the organoids returned to normal.
These exciting results demonstrate that correcting the specific defect in APC function can result in disease regression in animals that have colon cancer. Although no existing drugs are able to do this in patients, this work strongly suggests that further research to find such a drug may hold the promise of a more effective treatment for people who suffer from colon cancer.
Grant recipient in 2014-2016
Pancreatic cancer is an extremely lethal cancer for which no effective therapies are currently available. Marilyn Resh’s laboratory is focused on a molecular pathway controlled by Sonic hedgehog (Shh), a protein that contributes to pancreatic cancer development. Shh is produced by developing embryos. After birth, Shh expression is turned off and most normal adult tissues do not make the protein. However, in pancreatic cancer, Shh is aberrantly turned back on. This abnormal expression helps to promote the growth of pancreatic cancer. In order to function normally, Shh must be modified by having a fatty acid called palmitate attached to it. This process, known as palmitoylation, is mediated by a protein termed Hhat (Hedgehog palmitoyl acyltransferase). Dr. Resh and her colleagues hypothesized that it would be possible to block the action of Shh by inhibiting the ability of Hhat to attach palmitate to Shh.
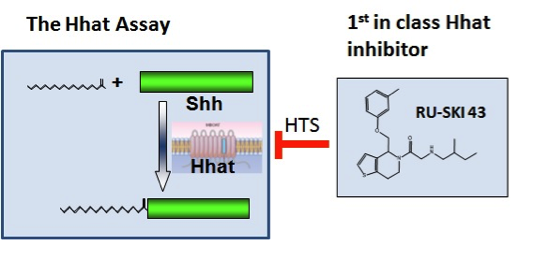
Using funds from the Geoffrey Beene Cancer Research Center, Dr. Resh set up a strategy to test her hypothesis and in doing so has developed new strategies to treat pancreatic cancer. In order to identify Hhat inhibitors, she established a method to measure Shh palmitoylation and used it to screen a library of 85,000 chemical compounds. This screen identified 95 compounds that directly inhibit Hhat; they chose the four best compounds for further study. Additional testing of one of the compounds, number 43, revealed that it enters cells, where it directly inhibits palmitoylation of Shh. Consistent with Dr. Resh’s hypothesis, compound 43 also inhibits the growth of human pancreatic cancer cells. These features support the concept that compound 43 can be developed into an effective drug for therapeutic treatment of pancreatic cancer in patients.
Stimulated by the success of her Geoffrey Beene–funded project, Dr. Resh and colleagues are continuing to develop agents that target Shh. In collaboration with the Tri-Institutional Therapeutics Discovery Institute and Takeda Pharmaceuticals, they are developing drugs that work as Hhat inhibitors. To date, no other studies have used Hhat as a target in pancreatic cancer (or any other cancer), making their approach innovative and unique. The Hhat inhibitors will be tested for their ability to block tumor growth in models of pancreatic, breast, and lung cancer in the laboratory, and ultimately in the clinic.
Chromosome rearrangements have been known for decades to be common in human cancers. These rearrangements often result in the fusion of genes, which alters protein function and contributes to cancer. As these fused genes are not normally present in a patient’s cells, drugs that inhibit their activity can eliminate cancer cells without harming normal tissues. Unfortunately, this type of mutation has proven very difficult to engineer in cells and even more so in model organisms, and this has greatly limited our ability to study them.
Funds from the Geoffrey Beene Cancer Research Center allowed Andrea Ventura to develop a procedure for generating virtually any chromosomal rearrangement in cells of adult mice and potentially other model organisms using a technique called genome editing. As a proof of concept for this approach, they generated a new mouse model of lung cancer caused by a chromosomal rearrangement that results in a gene fusion called EML4-ALK. They also showed that these tumors respond to chemical inhibitors of the fusion protein. Their innovative technology greatly expands our ability to make cancer models that accurately mimic the behavior of human tumors. Such models provide valuable preclinical platforms for evaluating new cancer drugs and testing ways to overcome drug resistance.